SnB Python API#
Literature#
We kindly ask that you cite the code and theory/method paper if you use ShakeNBreak in your work.
Preview: Mosquera-Lois, I.; Kavanagh, S. R. In Search of Hidden Defects, Matter 4 (8), 2602-2605, 2021
Code: Mosquera-Lois, I. & Kavanagh, S. R.; Walsh, A.; Scanlon, D. O. ShakeNBreak: Navigating the defect configurational landscape, Journal of Open Source Software 7 (80), 4817, 2022
Theory/Method: Mosquera-Lois, I. & Kavanagh, S. R.; Walsh, A.; Scanlon, D. O. Identifying the Ground State Structures of Defects in Solids, npj Comput Mater 9, 25 2023
News & Views: Mannodi-Kanakkithodi, A. The Devil is in the Defects, Nature Physics 2023 (Free-to-read link)
YouTube Overview (10 mins): ShakeNBreak: Symmetry-Breaking and Reconstruction at Defects in Solids
YouTube Seminar (35 mins): Seminar: Predicting the Atomic Structures of Defects
ShakeNBreak applied to Cd vacancies in CdTe (VCd)#
In this notebook we follow the full ShakeNBreak (SnB) workflow, where we:
Apply the defect distortions
Parse the geometry relaxation results
Re-generate any energy-lowering distortions found for some (but not all) charge states for a given defect
Plot the final energies to demonstrate what energy-lowering defect distortions have been identified
Then continue our defect calculations, confident we have obtained the ground-state structures.
Tip
ShakeNBreak can also be used to search for other energy-lowering distortions / localised species, such as polarons and self-trapped excitons. An example of using ShakeNBreak for this application is shown in the SnB Polarons Tutorial.
Table of contents#
Tip
You can run this notebook interactively through Google Colab or Binder using the launch buttons at the top of the docs tutorial page (click the Rocket ?? icon). If running on Colab, then you’ll need to run !pip install shakenbreak in a cell to install the package, and !git clone https://github.com/SMTG-Bham/shakenbreak to download the example data (and update paths in the code cells accordingly).
import os
import sys
import ase
import numpy as np
import pymatgen
import doped
from importlib_metadata import version
import shakenbreak
# Check versions
print("doped version:", version('doped') )
print("pymatgen version:", version('pymatgen') )
print("pymatgen-analysis-defects version:", version('pymatgen-analysis-defects') )
print("ase version:", version('ase') )
print("ShakeNBreak version:", version('shakenbreak') )
doped version: 2.3.0
pymatgen version: 2024.2.8
pymatgen-analysis-defects version: 2023.10.19
ase version: 3.22.1
ShakeNBreak version: 3.3.0
Rationale for SnB#
Note
Defect distortions often follow the change in electron count when introducing that defect to the system. For the neutral Cd vacancy (VCd0) for example, the removal of Cd and its two valence electrons means that local distortions are likely to involve two neighbouring Te atoms moving closer/further apart to accommodate the broken bonds. For the singly-charged vacancy, we are likely to have just one neighbouring Te moving, etc. This isn’t always the case, but typically points us in the right direction to search the PES, and has been confirmed to yield the best performance (see SI of Identifying the ground state structures of point defects in solids Mosquera-Lois, Kavanagh, Walsh and Scanlon 2022).
So, the SnB method involves distorting the initial bond lengths around the defect for a mesh of trial distortions, with the number of neighbours to distort dictated by the change in valence electron count, performing coarse Γ-only (vasp_gam) relaxations and then comparing the final energies, to see if we identify any lower energy defect structures.
1. Generate defects with doped/pymatgen#
The input defect objects for ShakeNBreak can be generated using either
doped, pymatgen or alternatively just
the bulk and defect structures can be provided. Below we show how to generate the defect python
objects using doped (Section
1.1) and pymatgen (Section 1.1).
In this case we take CdTe as our example system:
1.1 Generate defects with doped (recommended)#
To generate defects with doped, we can use the code below. This procedure is described in much more
detail on the doped docs, which also shows how to generate
and calculate the chemical potential terms (needed for defect formation energies), perform defect
supercell (finite-size) charge
corrections, calculate & plot the final defect formation energy diagram, and perform further defect
analysis.
from pymatgen.core.structure import Structure
from doped.generation import DefectsGenerator
bulk = Structure.from_file("../tests/data/vasp/CdTe/CdTe_Bulk_Primitive_POSCAR") # CdTe
defect_gen = DefectsGenerator(bulk)
Generating DefectEntry objects: 100.0%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| [00:12, 8.01it/s]
Vacancies Guessed Charges Conv. Cell Coords Wyckoff
----------- ----------------- ------------------- ---------
v_Cd [+1,0,-1,-2] [0.000,0.000,0.000] 4a
v_Te [+2,+1,0,-1] [0.250,0.250,0.250] 4c
Substitutions Guessed Charges Conv. Cell Coords Wyckoff
--------------- --------------------- ------------------- ---------
Cd_Te [+4,+3,+2,+1,0] [0.250,0.250,0.250] 4c
Te_Cd [+2,+1,0,-1,-2,-3,-4] [0.000,0.000,0.000] 4a
Interstitials Guessed Charges Conv. Cell Coords Wyckoff
--------------- --------------------- ------------------- ---------
Cd_i_C3v [+2,+1,0] [0.625,0.625,0.625] 16e
Cd_i_Td_Cd2.83 [+2,+1,0] [0.750,0.750,0.750] 4d
Cd_i_Td_Te2.83 [+2,+1,0] [0.500,0.500,0.500] 4b
Te_i_C3v [+4,+3,+2,+1,0,-1,-2] [0.625,0.625,0.625] 16e
Te_i_Td_Cd2.83 [+4,+3,+2,+1,0,-1,-2] [0.750,0.750,0.750] 4d
Te_i_Td_Te2.83 [+4,+3,+2,+1,0,-1,-2] [0.500,0.500,0.500] 4b
The number in the Wyckoff label is the site multiplicity/degeneracy of that defect in the conventional ('conv.') unit cell, which comprises 4 formula unit(s) of CdTe.
Note that Wyckoff letters can depend on the ordering of elements in the conventional standard structure, for which doped uses the spglib convention.
Tip
The DefectsGenerator object is highly-customisable (as are the other doped objects/functions):
DefectsGenerator?
Init signature:
DefectsGenerator(
structure: pymatgen.core.structure.Structure,
extrinsic: Union[str, List, Dict, NoneType] = None,
interstitial_coords: Optional[List] = None,
generate_supercell: bool = True,
charge_state_gen_kwargs: Optional[Dict] = None,
supercell_gen_kwargs: Optional[Dict] = None,
interstitial_gen_kwargs: Optional[Dict] = None,
target_frac_coords: Optional[List] = None,
processes: Optional[int] = None,
)
Docstring: Class for generating doped DefectEntry objects.
Init docstring:
Generates doped DefectEntry objects for defects in the input host
structure. By default, generates all intrinsic defects, but extrinsic
defects (impurities) can also be created using the ``extrinsic``
argument.
Interstitial sites are generated using Voronoi tessellation by default (found
to be the most reliable), which can be controlled using the
``interstitial_gen_kwargs`` argument (passed as keyword arguments to the
``VoronoiInterstitialGenerator`` class). Alternatively, a list of interstitial
sites (or single interstitial site) can be manually specified using the
``interstitial_coords`` argument.
By default, supercells are generated for each defect using the doped
``get_ideal_supercell_matrix()`` function (see docstring), with default settings
of ``min_image_distance = 10`` (minimum distance between periodic images of 10 Å),
``min_atoms = 50`` (minimum 50 atoms in the supercell) and ``ideal_threshold = 0.1``
(allow up to 10% larger supercell if it is a diagonal expansion of the primitive
or conventional cell). This uses a custom algorithm in ``doped`` to efficiently
search over possible supercell transformations and identify that with the minimum
number of atoms (hence computational cost) that satisfies the minimum image distance,
number of atoms and ``ideal_threshold`` constraints. These settings can be controlled
by specifying keyword arguments with ``supercell_gen_kwargs``, which are passed to
``get_ideal_supercell_matrix()`` (e.g. for a minimum image distance of 15 Å with at
least 100 atoms, use:
``supercell_gen_kwargs = {'min_image_distance': 15, 'min_atoms': 100}``). If the
input structure already satisfies these constraints (for the same number of atoms as
the ``doped``-generated supercell), then it will be used.
Alternatively if ``generate_supercell = False``, then no supercell is generated
and the input structure is used as the defect & bulk supercell. (Note this
may give a slightly different (but fully equivalent) set of coordinates).
The algorithm for determining defect entry names is to use the pymatgen defect
name (e.g. ``v_Cd``, ``Cd_Te`` etc.) for vacancies/antisites/substitutions, unless
there are multiple inequivalent sites for the defect, in which case the point
group of the defect site is appended (e.g. ``v_Cd_Td``, ``Cd_Te_Td`` etc.), and if
this is still not unique, then element identity and distance to the nearest
neighbour of the defect site is appended (e.g. ``v_Cd_Td_Te2.83``, ``Cd_Te_Td_Cd2.83``
etc.). For interstitials, the same naming scheme is used, but the point group
is always appended to the pymatgen defect name.
Possible charge states for the defects are estimated using the probability of
the corresponding defect element oxidation state, the magnitude of the charge
state, and the maximum magnitude of the host oxidation states (i.e. how
'charged' the host is), with large (absolute) charge states, low probability
oxidation states and/or greater charge/oxidation state magnitudes than that of
the host being disfavoured. This can be controlled using the
``probability_threshold`` (default = 0.0075) or ``padding`` (default = 1) keys in
the ``charge_state_gen_kwargs`` parameter, which are passed to the
``_charge_state_probability()`` function. The input and computed values used to
guess charge state probabilities are provided in the
``DefectEntry.charge_state_guessing_log`` attributes. See docs for examples of
modifying the generated charge states.
Args:
structure (Structure):
Structure of the host material (as a pymatgen Structure object).
If this is not the primitive unit cell, it will be reduced to the
primitive cell for defect generation, before supercell generation.
extrinsic (Union[str, List, Dict]):
List or dict of elements (or string for single element) to be used
for extrinsic defect generation (i.e. dopants/impurities). If a
list is provided, all possible substitutional defects for each
extrinsic element will be generated. If a dict is provided, the keys
should be the host elements to be substituted, and the values the
extrinsic element(s) to substitute in; as a string or list.
In both cases, all possible extrinsic interstitials are generated.
interstitial_coords (List):
List of fractional coordinates (corresponding to the input structure),
or a single set of fractional coordinates, to use as interstitial
defect site(s). Default (when interstitial_coords not specified) is
to automatically generate interstitial sites using Voronoi tessellation.
The input interstitial_coords are converted to
DefectsGenerator.prim_interstitial_coords, which are the corresponding
fractional coordinates in DefectsGenerator.primitive_structure (which
is used for defect generation), along with the multiplicity and
equivalent coordinates, sorted according to the doped convention.
generate_supercell (bool):
Whether to generate a supercell for the output defect entries
(using the custom algorithm in ``doped`` which efficiently searches over
possible supercell transformations and identifies that with the minimum
number of atoms (hence computational cost) that satisfies the minimum
image distance, number of atoms and ``ideal_threshold`` constraints
- which can be controlled with ``supercell_gen_kwargs``).
If False, then the input structure is used as the defect & bulk supercell.
(Note this may give a slightly different (but fully equivalent) set of coordinates).
charge_state_gen_kwargs (Dict):
Keyword arguments to be passed to the ``_charge_state_probability``
function (such as ``probability_threshold`` (default = 0.0075, used for
substitutions and interstitials) and ``padding`` (default = 1, used for
vacancies)) to control defect charge state generation.
supercell_gen_kwargs (Dict):
Keyword arguments to be passed to the ``get_ideal_supercell_matrix``
function (such as ``min_image_distance`` (default = 10), ``min_atoms``
(default = 50), ``ideal_threshold`` (default = 0.1), ``force_cubic``
- which enforces a (near-)cubic supercell output (default = False),
or ``force_diagonal`` (default = False)).
interstitial_gen_kwargs (Dict, bool):
Keyword arguments to be passed to the ``VoronoiInterstitialGenerator``
class (such as ``clustering_tol``, ``stol``, ``min_dist`` etc), or to
``InterstitialGenerator`` if ``interstitial_coords`` is specified.
If set to False, interstitial generation will be skipped entirely.
target_frac_coords (List):
Defects are placed at the closest equivalent site to these fractional
coordinates in the generated supercells. Default is [0.5, 0.5, 0.5]
if not set (i.e. the supercell centre, to aid visualisation).
processes (int):
Number of processes to use for multiprocessing. If not set, defaults to
one less than the number of CPUs available.
Attributes:
defect_entries (Dict): Dictionary of {defect_species: DefectEntry} for all
defect entries (with charge state and supercell properties) generated.
defects (Dict): Dictionary of {defect_type: [Defect, ...]} for all defect
objects generated.
primitive_structure (Structure): Primitive cell structure of the host
used to generate defects.
supercell_matrix (Matrix): Matrix to generate defect/bulk supercells from
the primitive cell structure.
bulk_supercell (Structure): Supercell structure of the host
(equal to primitive_structure * supercell_matrix).
conventional_structure (Structure): Conventional cell structure of the
host according to the Bilbao Crystallographic Server (BCS) definition,
used to determine defect site Wyckoff labels and multiplicities.
``DefectsGenerator`` input parameters are also set as attributes.
File: ~/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/doped/doped/generation.py
Type: type
Subclasses:
Note
As described on the doped tips docs page, if you have many possible interstitial sites (often the case in low-symmetry/multinary materials), then the recommended approach is to first perform Gamma-point-only relaxations (using vasp_gam) for the unperturbed neutral state of each generated interstitial candidate, then compare the energies of these trial neutral relaxations, and remove any candidates that either:
Are very high energy (>1 eV above the lowest energy site), and so are unlikely to form.
Relax to the same final structure/energy as other interstitial sites (despite different initial positions), and so are unnecessary to calculate.
1.2 Generate defects with pymatgen.analysis.defects#
Alternatively, we can also generate our defect objects directly with pymatgen.
You can skip ahead to Section 2 if you’re generating your
defects with doped as shown above.
Note
For a detailed guide on how to use pymatgen.analysis.defects to generate defects, see their tutorial.
A key point to note about their package is that defects are defined independently of the simulation cell
(e.g. the supercell). That is, they are generated using the Wigner-Seitz unit cell representation of
the bulk material. All information about a defect is captured with the Defect class. When information
about the simulation cell needs to be added, the DefectEntry object is used, as exemplified below.
from pymatgen.analysis.defects.core import Defect, Vacancy
from pymatgen.core.structure import Structure
# Using the _primitive_ cell of CdTe
bulk_primitive = Structure.from_file("../tests/data/vasp/CdTe/CdTe_Bulk_Primitive_POSCAR")
from pymatgen.analysis.defects.generators import VacancyGenerator
vac_gen = VacancyGenerator()
vacancies = vac_gen.get_defects(bulk_primitive)
print(vacancies)
[Cd Vacancy defect at site #0, Te Vacancy defect at site #1]
# Select Cd vacancy
v_Cd = vacancies[0]
v_Cd
Cd Vacancy defect at site #0
# We can specify the defect charge states like this:
v_Cd.user_charges = [-2, -1, 0]
After generating the pymatgen Defect object, we need to create a DefectEntry with a specified
defect supercell to use with SnB.
We can do this using the get_supercell_structure method, and the get_defect_entry_from_defect
function defined below:
from pymatgen.core.periodic_table import DummySpecies
max_atoms = 70
min_atoms = 30
min_length = 10
force_diagonal = False
defect_supercell = v_Cd.get_supercell_structure(
min_length=min_length, # in Angstrom
max_atoms=max_atoms,
min_atoms=min_atoms,
force_diagonal=force_diagonal,
dummy_species=DummySpecies("X"), # We use this to keep track of the frac coords of the
# defect in the supercell, but the dummy species will be removed in the next cell
)
print("Generated supercell:\n", defect_supercell.lattice)
Generated supercell:
13.086768 0.000000 0.000000
0.000000 13.086768 0.000000
0.000000 0.000000 13.086768
from pymatgen.analysis.defects.thermo import DefectEntry
from pymatgen.core.entries import ComputedStructureEntry
from pymatgen.core.periodic_table import DummySpecies
def get_defect_entry_from_defect(
defect: Defect,
defect_supercell: Structure,
charge_state: int,
dummy_species: DummySpecies=DummySpecies("X"),
):
"""Generate DefectEntry object from Defect object.
This is used to describe a Defect using a certain simulation cell.
"""
# Dummy species (used to keep track of the defect coords in the supercell)
# Find its fractional coordinates & remove it from supercell
dummy_site = [
site for site in defect_supercell
if site.species.elements[0].symbol == dummy_species.symbol
][0]
sc_defect_frac_coords = dummy_site.frac_coords
defect_supercell.remove(dummy_site)
computed_structure_entry = ComputedStructureEntry(
structure=defect_supercell,
energy=0.0, # needs to be set, so set to 0.0
)
return DefectEntry(
defect=defect,
charge_state=charge_state,
sc_entry=computed_structure_entry,
sc_defect_frac_coords=sc_defect_frac_coords,
)
defect_entry = get_defect_entry_from_defect(
defect=v_Cd,
charge_state=0,
defect_supercell=defect_supercell,
)
# Check the defect entry
print("Defect object stored as part of the DefectEntry:", defect_entry.defect)
Defect object stored as part of the DefectEntry: Cd Vacancy defect at site #0
print("Defect supercell stored as part of the DefectEntry:", defect_entry.sc_entry.structure)
Defect supercell stored as part of the DefectEntry: Full Formula (Cd31 Te32)
Reduced Formula: Cd31Te32
abc : 13.086768 13.086768 13.086768
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (63)
# SP a b c
--- ---- ----- ----- -----
0 Cd2+ 0.5 0 0
1 Cd2+ 0.75 0.25 0
2 Cd2+ 0.75 0 0.25
3 Cd2+ 0.25 0.25 0
4 Cd2+ 0.5 0.5 0
5 Cd2+ 0.75 0.75 0
6 Cd2+ 0.25 0 0.25
7 Cd2+ 0.5 0.25 0.25
8 Cd2+ 0.75 0.5 0.25
9 Cd2+ 0.5 0 0.5
10 Cd2+ 0.75 0.25 0.5
11 Cd2+ 0.75 0 0.75
12 Cd2+ 0 0.5 0
13 Cd2+ 0.25 0.75 0
14 Cd2+ 0 0.25 0.25
15 Cd2+ 0.25 0.5 0.25
16 Cd2+ 0.5 0.75 0.25
17 Cd2+ 0 0 0.5
18 Cd2+ 0.25 0.25 0.5
19 Cd2+ 0.5 0.5 0.5
20 Cd2+ 0.75 0.75 0.5
21 Cd2+ 0.25 0 0.75
22 Cd2+ 0.5 0.25 0.75
23 Cd2+ 0.75 0.5 0.75
24 Cd2+ 0 0.75 0.25
25 Cd2+ 0 0.5 0.5
26 Cd2+ 0.25 0.75 0.5
27 Cd2+ 0 0.25 0.75
28 Cd2+ 0.25 0.5 0.75
29 Cd2+ 0.5 0.75 0.75
30 Cd2+ 0 0.75 0.75
31 Te2- 0.625 0.125 0.875
32 Te2- 0.875 0.375 0.875
33 Te2- 0.875 0.125 0.125
34 Te2- 0.125 0.125 0.875
35 Te2- 0.375 0.375 0.875
36 Te2- 0.625 0.625 0.875
37 Te2- 0.875 0.875 0.875
38 Te2- 0.375 0.125 0.125
39 Te2- 0.625 0.375 0.125
40 Te2- 0.875 0.625 0.125
41 Te2- 0.625 0.125 0.375
42 Te2- 0.875 0.375 0.375
43 Te2- 0.875 0.125 0.625
44 Te2- 0.125 0.625 0.875
45 Te2- 0.375 0.875 0.875
46 Te2- 0.125 0.375 0.125
47 Te2- 0.375 0.625 0.125
48 Te2- 0.625 0.875 0.125
49 Te2- 0.125 0.125 0.375
50 Te2- 0.375 0.375 0.375
51 Te2- 0.625 0.625 0.375
52 Te2- 0.875 0.875 0.375
53 Te2- 0.375 0.125 0.625
54 Te2- 0.625 0.375 0.625
55 Te2- 0.875 0.625 0.625
56 Te2- 0.125 0.875 0.125
57 Te2- 0.125 0.625 0.375
58 Te2- 0.375 0.875 0.375
59 Te2- 0.125 0.375 0.625
60 Te2- 0.375 0.625 0.625
61 Te2- 0.625 0.875 0.625
62 Te2- 0.125 0.875 0.625
# We can also check the defect fractional coordinates in the supercell
print("Defect fractional coordinates in the supercell:", defect_entry.sc_defect_frac_coords)
Defect fractional coordinates in the supercell: [0. 0. 0.]
Important
Alternatively, if you have already generated your defect structure files with a different defects code, these can be directly fed to ShakeNBreak with the Distortions.from_structures() method as shown later on below.
2. Apply the SnB method to your defects#
Note
The default settings and parameter choices in this package have been tested and have performed best thus far (i.e. wider distortion ranges leading to the ground-state structure with lowest computational cost) – see SI of Identifying the ground state structures of point defects in solids.
If you encounter improved performance with non-default parameter choices, we’d love to know! Please get in touch via GitHub.
Tip
If you are investigating defects in hard/ionic/magnetic/correlated materials, or systems involving spectator ions (like A in ABX3), there are some extra considerations for boosting the performance & efficiency of SnB listed on the Miscellaneous Tips & Tricks docs page.
2.1 Generating distorted structures#
from shakenbreak.input import Distortions
# In order to determine the number of the defect nearest neighbours to distort (based on the change
# in valence electrons mentioned above), SnB uses the oxidation states of atoms in our material:
# If not specified, the code will guess these, otherwise you can specify as such:
# oxidation_states = {"Cd": +2, "Te": -2} # specify atom oxidation states
# Create an instance of Distortion class with the defects and distortion parameters
# If distortion parameters are not specified, the default values are used
Dist = Distortions(defect_gen) # initialise with doped DefectsGenerator
Oxidation states were not explicitly set, thus have been guessed as {'Cd': 2.0, 'Te': -2.0}. If this is unreasonable you should manually set oxidation_states
The Distortions class is flexible to the user input, so can take a doped DefectsGenerator
object (shown in Section 1.1), a list of DefectEntrys, or a dictionary of DefectEntrys (in which
case the dictionary keys are used as the defect names), or a single DefectEntry as inputs.
Alternatively, Distortions can be initialised directly from pymatgen structures of the bulk and
defect supercell, using the from_structures() class method, or from structure files (e.g. POSCARs)
on the CLI using snb-generate – though
in general the python API route shown here is preferred as it is more efficient and offers more control.
These possibilities as well as the optional distortion parameters are detailed in the Distortions class docstring:
Distortions?
# We can check the distortion parameters using some of the class properties
print(f"Bond distortions: {Dist.bond_distortions}")
print(f"Rattle standard deviation: {Dist.stdev:.2f} Å") # set to 10% of the bulk bond length by default, typically a reasonable value
As mentioned above, we can also initialise Distortions directly from our pre-generated defect structures, using the Distortions.from_structures() method like this:
from pymatgen.core.structure import Structure
V_Cd_struc = Structure.from_file("../tests/data/vasp/CdTe/CdTe_V_Cd_POSCAR")
bulk_struc = Structure.from_file("../tests/data/vasp/CdTe/CdTe_Bulk_Supercell_POSCAR")
Dist = Distortions.from_structures(defects = V_Cd_struc, bulk = bulk_struc)
Defect charge states will be set to the range: 0 – {Defect oxidation state}, with a `padding = 1` on either side of this range.
Oxidation states were not explicitly set, thus have been guessed as {'Cd': 2.0, 'Te': -2.0}. If this is unreasonable you should manually set oxidation_states
Tip
You can restrict the ions that are distorted to a certain element using the keyword distorted_elements.
We can check it using the class attribute:
print("User defined elements to distort:", Dist.distorted_elements)
User defined elements to distort: None
If None, it means no restrictions, so nearest neighbours are distorted (recommended default,
unless you have reason to suspect otherwise; see Tips )
If we’re only interested in generating distorted structures, but not in writing VASP/other codes input files, we can use the class method Distortions.apply_distortions() to do this.
defects_dict, distortion_metadata = Dist.apply_distortions()
Applying ShakeNBreak... Will apply the following bond distortions: ['-0.6', '-0.5', '-0.4', '-0.3', '-0.2', '-0.1', '0.0', '0.1', '0.2', '0.3', '0.4', '0.5', '0.6']. Then, will rattle with a std dev of 0.28 Å
Defect: v_Cd_Td_Te2.83
Number of missing electrons in neutral state: 2
Defect v_Cd_Td_Te2.83 in charge state: -3. Number of distorted neighbours: 1
Defect v_Cd_Td_Te2.83 in charge state: -2. Number of distorted neighbours: 0
Defect v_Cd_Td_Te2.83 in charge state: -1. Number of distorted neighbours: 1
Defect v_Cd_Td_Te2.83 in charge state: 0. Number of distorted neighbours: 2
Defect v_Cd_Td_Te2.83 in charge state: +1. Number of distorted neighbours: 3
The output dictionary contains information about each defect:
print("Keys for each defect entry:", defects_dict["v_Cd_Td_Te2.83"].keys())
Keys for each defect entry: dict_keys(['defect_type', 'defect_site', 'defect_supercell_site', 'charges'])
As well as the distorted structures for each charge state of all defects. We can access the distorted structures of v_Cd_0 like this:
print("\nUndistorted and distorted structures:")
defects_dict["v_Cd_Td_Te2.83"]["charges"][0]["structures"]
Undistorted and distorted structures:
{'Unperturbed': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd (Cd2+) (0.0, 0.0, 6.543) [0.0, 0.0, 0.5]
PeriodicSite: Cd (Cd2+) (0.0, 6.543, 0.0) [0.0, 0.5, 0.0]
PeriodicSite: Cd (Cd2+) (0.0, 6.543, 6.543) [0.0, 0.5, 0.5]
PeriodicSite: Cd (Cd2+) (6.543, 0.0, 0.0) [0.5, 0.0, 0.0]
PeriodicSite: Cd (Cd2+) (6.543, 0.0, 6.543) [0.5, 0.0, 0.5]
PeriodicSite: Cd (Cd2+) (6.543, 6.543, 0.0) [0.5, 0.5, 0.0]
PeriodicSite: Cd (Cd2+) (6.543, 6.543, 6.543) [0.5, 0.5, 0.5]
PeriodicSite: Cd (Cd2+) (0.0, 3.272, 3.272) [0.0, 0.25, 0.25]
PeriodicSite: Cd (Cd2+) (0.0, 3.272, 9.815) [0.0, 0.25, 0.75]
PeriodicSite: Cd (Cd2+) (0.0, 9.815, 3.272) [0.0, 0.75, 0.25]
PeriodicSite: Cd (Cd2+) (0.0, 9.815, 9.815) [0.0, 0.75, 0.75]
PeriodicSite: Cd (Cd2+) (6.543, 3.272, 3.272) [0.5, 0.25, 0.25]
PeriodicSite: Cd (Cd2+) (6.543, 3.272, 9.815) [0.5, 0.25, 0.75]
PeriodicSite: Cd (Cd2+) (6.543, 9.815, 3.272) [0.5, 0.75, 0.25]
PeriodicSite: Cd (Cd2+) (6.543, 9.815, 9.815) [0.5, 0.75, 0.75]
PeriodicSite: Cd (Cd2+) (3.272, 0.0, 3.272) [0.25, 0.0, 0.25]
PeriodicSite: Cd (Cd2+) (3.272, 0.0, 9.815) [0.25, 0.0, 0.75]
PeriodicSite: Cd (Cd2+) (3.272, 6.543, 3.272) [0.25, 0.5, 0.25]
PeriodicSite: Cd (Cd2+) (3.272, 6.543, 9.815) [0.25, 0.5, 0.75]
PeriodicSite: Cd (Cd2+) (9.815, 0.0, 3.272) [0.75, 0.0, 0.25]
PeriodicSite: Cd (Cd2+) (9.815, 0.0, 9.815) [0.75, 0.0, 0.75]
PeriodicSite: Cd (Cd2+) (9.815, 6.543, 3.272) [0.75, 0.5, 0.25]
PeriodicSite: Cd (Cd2+) (9.815, 6.543, 9.815) [0.75, 0.5, 0.75]
PeriodicSite: Cd (Cd2+) (3.272, 3.272, 0.0) [0.25, 0.25, 0.0]
PeriodicSite: Cd (Cd2+) (3.272, 3.272, 6.543) [0.25, 0.25, 0.5]
PeriodicSite: Cd (Cd2+) (3.272, 9.815, 0.0) [0.25, 0.75, 0.0]
PeriodicSite: Cd (Cd2+) (3.272, 9.815, 6.543) [0.25, 0.75, 0.5]
PeriodicSite: Cd (Cd2+) (9.815, 3.272, 0.0) [0.75, 0.25, 0.0]
PeriodicSite: Cd (Cd2+) (9.815, 3.272, 6.543) [0.75, 0.25, 0.5]
PeriodicSite: Cd (Cd2+) (9.815, 9.815, 0.0) [0.75, 0.75, 0.0]
PeriodicSite: Cd (Cd2+) (9.815, 9.815, 6.543) [0.75, 0.75, 0.5]
PeriodicSite: Te (Te2-) (1.636, 1.636, 4.908) [0.125, 0.125, 0.375]
PeriodicSite: Te (Te2-) (1.636, 1.636, 11.45) [0.125, 0.125, 0.875]
PeriodicSite: Te (Te2-) (1.636, 8.179, 4.908) [0.125, 0.625, 0.375]
PeriodicSite: Te (Te2-) (1.636, 8.179, 11.45) [0.125, 0.625, 0.875]
PeriodicSite: Te (Te2-) (8.179, 1.636, 4.908) [0.625, 0.125, 0.375]
PeriodicSite: Te (Te2-) (8.179, 1.636, 11.45) [0.625, 0.125, 0.875]
PeriodicSite: Te (Te2-) (8.179, 8.179, 4.908) [0.625, 0.625, 0.375]
PeriodicSite: Te (Te2-) (8.179, 8.179, 11.45) [0.625, 0.625, 0.875]
PeriodicSite: Te (Te2-) (1.636, 4.908, 1.636) [0.125, 0.375, 0.125]
PeriodicSite: Te (Te2-) (1.636, 4.908, 8.179) [0.125, 0.375, 0.625]
PeriodicSite: Te (Te2-) (1.636, 11.45, 1.636) [0.125, 0.875, 0.125]
PeriodicSite: Te (Te2-) (1.636, 11.45, 8.179) [0.125, 0.875, 0.625]
PeriodicSite: Te (Te2-) (8.179, 4.908, 1.636) [0.625, 0.375, 0.125]
PeriodicSite: Te (Te2-) (8.179, 4.908, 8.179) [0.625, 0.375, 0.625]
PeriodicSite: Te (Te2-) (8.179, 11.45, 1.636) [0.625, 0.875, 0.125]
PeriodicSite: Te (Te2-) (8.179, 11.45, 8.179) [0.625, 0.875, 0.625]
PeriodicSite: Te (Te2-) (4.908, 1.636, 1.636) [0.375, 0.125, 0.125]
PeriodicSite: Te (Te2-) (4.908, 1.636, 8.179) [0.375, 0.125, 0.625]
PeriodicSite: Te (Te2-) (4.908, 8.179, 1.636) [0.375, 0.625, 0.125]
PeriodicSite: Te (Te2-) (4.908, 8.179, 8.179) [0.375, 0.625, 0.625]
PeriodicSite: Te (Te2-) (11.45, 1.636, 1.636) [0.875, 0.125, 0.125]
PeriodicSite: Te (Te2-) (11.45, 1.636, 8.179) [0.875, 0.125, 0.625]
PeriodicSite: Te (Te2-) (11.45, 8.179, 1.636) [0.875, 0.625, 0.125]
PeriodicSite: Te (Te2-) (11.45, 8.179, 8.179) [0.875, 0.625, 0.625]
PeriodicSite: Te (Te2-) (4.908, 4.908, 4.908) [0.375, 0.375, 0.375]
PeriodicSite: Te (Te2-) (4.908, 4.908, 11.45) [0.375, 0.375, 0.875]
PeriodicSite: Te (Te2-) (4.908, 11.45, 4.908) [0.375, 0.875, 0.375]
PeriodicSite: Te (Te2-) (4.908, 11.45, 11.45) [0.375, 0.875, 0.875]
PeriodicSite: Te (Te2-) (11.45, 4.908, 4.908) [0.875, 0.375, 0.375]
PeriodicSite: Te (Te2-) (11.45, 4.908, 11.45) [0.875, 0.375, 0.875]
PeriodicSite: Te (Te2-) (11.45, 11.45, 4.908) [0.875, 0.875, 0.375]
PeriodicSite: Te (Te2-) (11.45, 11.45, 11.45) [0.875, 0.875, 0.875],
'distortions': {'Bond_Distortion_-60.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (0.6543, 0.6543, 12.43) [0.05, 0.05, 0.95]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (0.6543, 12.43, 0.6543) [0.05, 0.95, 0.05]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_-50.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (0.8179, 0.8179, 12.27) [0.0625, 0.0625, 0.9375]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (0.8179, 12.27, 0.8179) [0.0625, 0.9375, 0.0625]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_-40.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (0.9815, 0.9815, 12.11) [0.075, 0.075, 0.925]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (0.9815, 12.11, 0.9815) [0.075, 0.925, 0.075]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_-30.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (1.145, 1.145, 11.94) [0.0875, 0.0875, 0.9125]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (1.145, 11.94, 1.145) [0.0875, 0.9125, 0.0875]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_-20.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (1.309, 1.309, 11.78) [0.1, 0.1, 0.9]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (1.309, 11.78, 1.309) [0.1, 0.9, 0.1]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_-10.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (1.472, 1.472, 11.61) [0.1125, 0.1125, 0.8875]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (1.472, 11.61, 1.472) [0.1125, 0.8875, 0.1125]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_0.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (1.636, 1.636, 11.45) [0.125, 0.125, 0.875]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (1.636, 11.45, 1.636) [0.125, 0.875, 0.125]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_10.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (1.799, 1.799, 11.29) [0.1375, 0.1375, 0.8625]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (1.799, 11.29, 1.799) [0.1375, 0.8625, 0.1375]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_20.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (1.963, 1.963, 11.12) [0.15, 0.15, 0.85]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (1.963, 11.12, 1.963) [0.15, 0.85, 0.15]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_30.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (2.127, 2.127, 10.96) [0.1625, 0.1625, 0.8375]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (2.127, 10.96, 2.127) [0.1625, 0.8375, 0.1625]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_40.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (2.29, 2.29, 10.8) [0.175, 0.175, 0.825]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (2.29, 10.8, 2.29) [0.175, 0.825, 0.175]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_50.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (2.454, 2.454, 10.63) [0.1875, 0.1875, 0.8125]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (2.454, 10.63, 2.454) [0.1875, 0.8125, 0.1875]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067],
'Bond_Distortion_60.0%': Structure Summary
Lattice
abc : 13.086768 13.086768 13.086768
angles : 90.0 90.0 90.0
volume : 2241.2856479961474
A : 13.086768 0.0 0.0
B : 0.0 13.086768 0.0
C : 0.0 0.0 13.086768
pbc : True True True
PeriodicSite: Cd2+ (0.08958, 0.2795, 6.5) [0.006845, 0.02136, 0.4967]
PeriodicSite: Cd2+ (0.04022, 6.923, -0.04308) [0.003074, 0.529, -0.003292]
PeriodicSite: Cd2+ (-0.1846, 6.619, 6.332) [-0.0141, 0.5058, 0.4839]
PeriodicSite: Cd2+ (6.625, 0.2106, -0.3562) [0.5063, 0.01609, -0.02722]
PeriodicSite: Cd2+ (6.471, 0.2184, 6.124) [0.4945, 0.01669, 0.468]
PeriodicSite: Cd2+ (7.055, 6.707, -0.1046) [0.5391, 0.5125, -0.007992]
PeriodicSite: Cd2+ (6.401, 6.223, 6.427) [0.4891, 0.4755, 0.4911]
PeriodicSite: Cd2+ (0.02002, 3.176, 3.109) [0.00153, 0.2427, 0.2375]
PeriodicSite: Cd2+ (0.08734, 3.3, 9.288) [0.006674, 0.2521, 0.7097]
PeriodicSite: Cd2+ (0.1104, 9.485, 3.089) [0.008438, 0.7248, 0.2361]
PeriodicSite: Cd2+ (-0.2325, 9.461, 9.953) [-0.01776, 0.7229, 0.7606]
PeriodicSite: Cd2+ (6.479, 3.319, 3.089) [0.4951, 0.2536, 0.236]
PeriodicSite: Cd2+ (6.722, 3.356, 9.805) [0.5136, 0.2565, 0.7492]
PeriodicSite: Cd2+ (6.441, 9.742, 3.401) [0.4922, 0.7444, 0.2599]
PeriodicSite: Cd2+ (6.906, 9.638, 9.544) [0.5277, 0.7364, 0.7293]
PeriodicSite: Cd2+ (3.394, -0.1058, 3.65) [0.2593, -0.008082, 0.2789]
PeriodicSite: Cd2+ (3.462, 0.111, 9.784) [0.2645, 0.008485, 0.7476]
PeriodicSite: Cd2+ (3.57, 6.257, 3.183) [0.2728, 0.4781, 0.2433]
PeriodicSite: Cd2+ (3.019, 6.647, 10.2) [0.2307, 0.5079, 0.7795]
PeriodicSite: Cd2+ (9.342, 0.03649, 2.872) [0.7139, 0.002789, 0.2195]
PeriodicSite: Cd2+ (10.09, 0.1205, 10.08) [0.7707, 0.009206, 0.7703]
PeriodicSite: Cd2+ (9.618, 6.663, 3.423) [0.7349, 0.5092, 0.2616]
PeriodicSite: Cd2+ (9.638, 6.435, 9.48) [0.7365, 0.4917, 0.7244]
PeriodicSite: Cd2+ (3.655, 3.197, -0.3796) [0.2793, 0.2443, -0.029]
PeriodicSite: Cd2+ (3.717, 3.562, 6.305) [0.2841, 0.2722, 0.4818]
PeriodicSite: Cd2+ (3.218, 10.3, 0.1662) [0.2459, 0.787, 0.0127]
PeriodicSite: Cd2+ (3.325, 9.973, 6.478) [0.2541, 0.7621, 0.495]
PeriodicSite: Cd2+ (9.786, 3.454, 0.3456) [0.7477, 0.2639, 0.02641]
PeriodicSite: Cd2+ (10.5, 3.528, 7.021) [0.8022, 0.2696, 0.5365]
PeriodicSite: Cd2+ (9.788, 9.739, 0.04483) [0.7479, 0.7442, 0.003425]
PeriodicSite: Cd2+ (9.935, 9.765, 6.804) [0.7591, 0.7461, 0.5199]
PeriodicSite: Te2- (1.547, 1.98, 5.395) [0.1182, 0.1513, 0.4122]
PeriodicSite: Te2- (2.617, 2.617, 10.47) [0.2, 0.2, 0.8]
PeriodicSite: Te2- (1.693, 7.598, 4.615) [0.1294, 0.5806, 0.3526]
PeriodicSite: Te2- (1.136, 8.308, 11.55) [0.08677, 0.6349, 0.8828]
PeriodicSite: Te2- (7.889, 2.169, 4.841) [0.6028, 0.1657, 0.3699]
PeriodicSite: Te2- (8.276, 2.219, 11.29) [0.6324, 0.1696, 0.8626]
PeriodicSite: Te2- (8.204, 8.164, 4.989) [0.6269, 0.6238, 0.3812]
PeriodicSite: Te2- (8.314, 8.195, 11.47) [0.6353, 0.6262, 0.8767]
PeriodicSite: Te2- (2.015, 4.642, 2.096) [0.154, 0.3547, 0.1602]
PeriodicSite: Te2- (1.687, 4.993, 8.336) [0.1289, 0.3816, 0.637]
PeriodicSite: Te2- (2.617, 10.47, 2.617) [0.2, 0.8, 0.2]
PeriodicSite: Te2- (1.406, 11.77, 8.203) [0.1075, 0.8993, 0.6268]
PeriodicSite: Te2- (8.13, 4.949, 1.534) [0.6213, 0.3781, 0.1172]
PeriodicSite: Te2- (8.387, 4.59, 7.866) [0.6409, 0.3507, 0.601]
PeriodicSite: Te2- (7.817, 11.61, 1.649) [0.5973, 0.8874, 0.126]
PeriodicSite: Te2- (8.217, 11.68, 8.444) [0.6279, 0.8927, 0.6452]
PeriodicSite: Te2- (5.052, 1.411, 1.871) [0.386, 0.1078, 0.143]
PeriodicSite: Te2- (4.149, 1.799, 7.797) [0.317, 0.1375, 0.5958]
PeriodicSite: Te2- (4.53, 8.048, 1.479) [0.3461, 0.615, 0.113]
PeriodicSite: Te2- (4.657, 7.939, 7.666) [0.3559, 0.6066, 0.5858]
PeriodicSite: Te2- (11.27, 2.077, 1.94) [0.8608, 0.1587, 0.1482]
PeriodicSite: Te2- (11.39, 1.811, 8.485) [0.8706, 0.1384, 0.6484]
PeriodicSite: Te2- (11.58, 8.432, 1.116) [0.8846, 0.6443, 0.08528]
PeriodicSite: Te2- (11.41, 7.983, 8.342) [0.8716, 0.61, 0.6375]
PeriodicSite: Te2- (4.809, 4.54, 4.531) [0.3674, 0.3469, 0.3463]
PeriodicSite: Te2- (5.258, 4.554, 11.69) [0.4018, 0.348, 0.893]
PeriodicSite: Te2- (4.66, 11.47, 5.087) [0.3561, 0.8763, 0.3887]
PeriodicSite: Te2- (4.764, 11.6, 11.67) [0.3641, 0.8867, 0.8921]
PeriodicSite: Te2- (11.69, 4.757, 4.892) [0.893, 0.3635, 0.3738]
PeriodicSite: Te2- (11.72, 5.1, 11.69) [0.8956, 0.3897, 0.8936]
PeriodicSite: Te2- (11.83, 11.33, 4.9) [0.9043, 0.8659, 0.3744]
PeriodicSite: Te2- (11.36, 10.99, 11.87) [0.8677, 0.8399, 0.9067]}}
(If you are viewing this on the SnB Python API tutorial docs page, long output cells like this and printed dictionaries/structures below are scrollable!)
2.2 Generating VASP input files for the distorted structures#
If we want to generate VASP input files, we can use the class method Distortions.write_vasp_files() (instead of Distortions.apply_distortions())
# using the recommended doped defect generation approach:
Dist = Distortions(defect_gen) # not needed if you've already initialised Distortions with doped above
defects_dict, distortion_metadata = Dist.write_vasp_files()
Oxidation states were not explicitly set, thus have been guessed as {'Cd': 2.0, 'Te': -2.0}. If this is unreasonable you should manually set oxidation_states
Applying ShakeNBreak... Will apply the following bond distortions: ['-0.6', '-0.5', '-0.4', '-0.3', '-0.2', '-0.1', '0.0', '0.1', '0.2', '0.3', '0.4', '0.5', '0.6']. Then, will rattle with a std dev of 0.28 Å
Defect: v_Cd
Number of missing electrons in neutral state: 2
Defect v_Cd in charge state: +1. Number of distorted neighbours: 3
Defect v_Cd in charge state: 0. Number of distorted neighbours: 2
Defect v_Cd in charge state: -1. Number of distorted neighbours: 1
Defect v_Cd in charge state: -2. Number of distorted neighbours: 0
Defect: v_Te
Number of extra electrons in neutral state: 2
Defect v_Te in charge state: +2. Number of distorted neighbours: 0
Defect v_Te in charge state: +1. Number of distorted neighbours: 1
Defect v_Te in charge state: 0. Number of distorted neighbours: 2
Defect v_Te in charge state: -1. Number of distorted neighbours: 3
Defect: Cd_Te
Number of extra electrons in neutral state: 4
Defect Cd_Te in charge state: +4. Number of distorted neighbours: 0
Defect Cd_Te in charge state: +3. Number of distorted neighbours: 1
Defect Cd_Te in charge state: +2. Number of distorted neighbours: 2
Defect Cd_Te in charge state: +1. Number of distorted neighbours: 3
Defect Cd_Te in charge state: 0. Number of distorted neighbours: 4
Defect: Te_Cd
Number of missing electrons in neutral state: 4
Defect Te_Cd in charge state: +2. Number of distorted neighbours: 2
Defect Te_Cd in charge state: +1. Number of distorted neighbours: 3
Defect Te_Cd in charge state: 0. Number of distorted neighbours: 4
Defect Te_Cd in charge state: -1. Number of distorted neighbours: 3
Defect Te_Cd in charge state: -2. Number of distorted neighbours: 2
Defect Te_Cd in charge state: -3. Number of distorted neighbours: 1
Defect Te_Cd in charge state: -4. Number of distorted neighbours: 0
Defect: Cd_i_C3v
Number of extra electrons in neutral state: 2
Defect Cd_i_C3v in charge state: +2. Number of distorted neighbours: 0
Defect Cd_i_C3v in charge state: +1. Number of distorted neighbours: 1
Defect Cd_i_C3v in charge state: 0. Number of distorted neighbours: 2
Defect: Cd_i_Td_Cd2.83
Number of extra electrons in neutral state: 2
Defect Cd_i_Td_Cd2.83 in charge state: +2. Number of distorted neighbours: 0
Defect Cd_i_Td_Cd2.83 in charge state: +1. Number of distorted neighbours: 1
Defect Cd_i_Td_Cd2.83 in charge state: 0. Number of distorted neighbours: 2
Defect: Cd_i_Td_Te2.83
Number of extra electrons in neutral state: 2
Defect Cd_i_Td_Te2.83 in charge state: +2. Number of distorted neighbours: 0
Defect Cd_i_Td_Te2.83 in charge state: +1. Number of distorted neighbours: 1
Defect Cd_i_Td_Te2.83 in charge state: 0. Number of distorted neighbours: 2
Defect: Te_i_C3v
Number of missing electrons in neutral state: 2
Defect Te_i_C3v in charge state: +4. Number of distorted neighbours: 2
Defect Te_i_C3v in charge state: +3. Number of distorted neighbours: 3
Defect Te_i_C3v in charge state: +2. Number of distorted neighbours: 4
Defect Te_i_C3v in charge state: +1. Number of distorted neighbours: 3
Defect Te_i_C3v in charge state: 0. Number of distorted neighbours: 2
Defect Te_i_C3v in charge state: -1. Number of distorted neighbours: 1
Defect Te_i_C3v in charge state: -2. Number of distorted neighbours: 0
Defect: Te_i_Td_Cd2.83
Number of missing electrons in neutral state: 2
Defect Te_i_Td_Cd2.83 in charge state: +4. Number of distorted neighbours: 2
Defect Te_i_Td_Cd2.83 in charge state: +3. Number of distorted neighbours: 3
Defect Te_i_Td_Cd2.83 in charge state: +2. Number of distorted neighbours: 4
Defect Te_i_Td_Cd2.83 in charge state: +1. Number of distorted neighbours: 3
Defect Te_i_Td_Cd2.83 in charge state: 0. Number of distorted neighbours: 2
Defect Te_i_Td_Cd2.83 in charge state: -1. Number of distorted neighbours: 1
Defect Te_i_Td_Cd2.83 in charge state: -2. Number of distorted neighbours: 0
Defect: Te_i_Td_Te2.83
Number of missing electrons in neutral state: 2
Defect Te_i_Td_Te2.83 in charge state: +4. Number of distorted neighbours: 2
Defect Te_i_Td_Te2.83 in charge state: +3. Number of distorted neighbours: 3
Defect Te_i_Td_Te2.83 in charge state: +2. Number of distorted neighbours: 4
Defect Te_i_Td_Te2.83 in charge state: +1. Number of distorted neighbours: 3
Defect Te_i_Td_Te2.83 in charge state: 0. Number of distorted neighbours: 2
Defect Te_i_Td_Te2.83 in charge state: -1. Number of distorted neighbours: 1
Defect Te_i_Td_Te2.83 in charge state: -2. Number of distorted neighbours: 0
Note
Using the user_incar_settings optional argument for Distortions.write_vasp_files() above, we can also specify some custom INCAR tags to match our converged ENCUT for this system and optimal NCORE for the HPC we will run the calculations on. Note that any INCAR flags that aren’t numbers (e.g. {"IBRION": 1}) or True/False (e.g. {"LREAL": False}) need to be input as strings with quotation marks (e.g. {"ALGO": "All"}).
For the recommended default coarse structure-searching INCAR settings, either have a look at the incar.yaml file in the SnB_input_files folder or at the generated files:
!cat ./Te_i_Td_Te2.83_0/Bond_Distortion_-10.0%/INCAR
# May want to change NCORE, KPAR, AEXX, ENCUT, IBRION, LREAL, NUPDOWN, ISPIN = Typical variable parameters
# ShakeNBreak INCAR with coarse settings to maximise speed with sufficient accuracy for qualitative structure searching =
AEXX = 0.25
ALGO = Normal # change to all if zhegv, fexcp/f or zbrent errors encountered (done automatically by snb-run)
EDIFF = 1e-05
EDIFFG = -0.01
ENCUT = 300
GGA = Pe
HFSCREEN = 0.208
IBRION = 2
ICHARG = 1
ICORELEVEL = 0 # needed if using the kumagai-oba (efnv) anisotropic charge correction scheme
ISIF = 2
ISMEAR = 0
ISPIN = 2
ISYM = 0 # symmetry breaking extremely likely for defects
LASPH = True
LCHARG = False
LHFCALC = True
LMAXMIX = 4
LORBIT = 11
LREAL = Auto
LVHAR = True
LWAVE = False
NCORE = 16
NEDOS = 2000
NELECT = 492.0
NELM = 40
NSW = 300
NUPDOWN = 0
PREC = Accurate
PRECFOCK = Fast
ROPT = 1e-3 1e-3
SIGMA = 0.05
Note
Note that the NELECT INCAR tag (number of electrons) is automatically determined based on the choice
of POTCARs. The default in ShakeNBreak (and doped) is to use the
MPRelaxSet POTCAR choices, but if you’re using
different ones, make sure to set user_potcar_settings in write_vasp_files(), so that NELECT is then set
accordingly.
This requires the pymatgen config file $HOME/.pmgrc.yaml to be properly set up as detailed on the Installation docs page.
More information on the distortions generated can be obtained by setting verbose = True.
Note
For generating the input files for other electronic structure codes (Quantum Espresso, FHI-aims, CP2K, CASTEP), see the (Optional) Generate input files for other codes section at the end of this notebook.
3. Send to HPCs and run relaxations#
The next step is to send the generated input files to our HPC and run the relaxation calculations.
We can use the snb-run CLI function to efficiently and automaticaly run and manage the calculations;
see the Submitting the geometry optimisations section of the CLI tutorial for this.
4. Plot energies vs distortion#
To see if SnB found any energy-lowering distortions, we can plot the results using the functions in shakenbreak.plotting.
Here we can either perform this analysis using the snb CLI commands on our HPC, as described in the
Analysis & Plotting docs page, or using
the python API workflow described below:
a) For VASP users:#
Parse the energies obtained by running the snb-parse command from the top-level folder containing your defect folders (e.g. v_Cd_0 etc. (with subfolders: v_Cd_0/Bond_Distortion_10.0% etc.)). This will parse the energies and store them in a v_Cd_0.yaml etc file in the defect folders, to allow easy plotting and analysis.
It is also recommended to parse the final structures (CONTCARs files if using VASP) obtained with each distortion relaxation for further structural analysis, which is done automatically when downloaded to your local folders as below.
Copying these data to your local PC can be done quickly from your local folder top-level folder (containing v_Cd_0 etc) with the following code:
shopt -s extglob # ensure extended globbing (pattern matching) is enabled
for defect in ./*{_,_-}[0-9]/; do cd $defect;
scp {remote_machine}:{path to ShakeNBreak folders}/${defect}${defect%?}.yaml .;
for distortion in (Bond_Distortion|Unperturbed|Rattled)*/;
do scp {remote_machine}:{path to ShakeNBreak folders}/${defect}${distortion}CONTCAR ${distortion};
done; cd ..; done
making sure to change {remote_machine} and {path to ShakeNBreak folders} to the correct values in your case.
Note
If you’re using MacOS (i.e. zsh shell) you may need to run this command first for this loop to work:
setopt +o nomatch
b) If using CP2K, Quantum Espresso, CASTEP or FHI-aims:#
Parse the energies obtained by running the snb-parse command from the top-level folder containing your defect folders (e.g. v_Cd_0 etc. (with subfolders: v_Cd_0/Bond_Distortion_10.0% etc.)) and setting the --code option (e.g. snb-parse --code cp2k). This will parse the energies and store them in a v_Cd_0.yaml etc file in the defect folders, to allow easy plotting and analysis.
It is also recommended to parse the final structures obtained with each relaxation for further structural analysis. Depending on the code the structure information is read from:
CP2K: restart fileQuantum Espresso: output fileCASTEP: output file (i.e..castep)FHI-aims: output file
For demonstration purposes in this example notebook, we’ll use some (fake) example data: (Don’t do this if you’re actually running the calculations and have downloaded the data as instructed above)
!rm -r ./*Cd_*/
!rm -r ./*Te_*/
!cp -r ../tests/data/example_results/v_Cd_* .
# may need to change path if you've moved the example notebook elsewhere
from shakenbreak import energy_lowering_distortions, plotting
defect_charges_dict = energy_lowering_distortions.read_defects_directories()
low_energy_defects = energy_lowering_distortions.get_energy_lowering_distortions(defect_charges_dict)
v_Cd
v_Cd_0: Energy difference between minimum, found with -0.6 bond distortion, and unperturbed: -0.76 eV.
Energy lowering distortion found for v_Cd with charge 0. Adding to low_energy_defects dictionary.
v_Cd_-1: Energy difference between minimum, found with 0.2 bond distortion, and unperturbed: -0.90 eV.
New (according to structure matching) low-energy distorted structure found for v_Cd_-1, adding to low_energy_defects['v_Cd'] list.
No energy lowering distortion with energy difference greater than min_e_diff = 0.05 eV found for v_Cd with charge -2.
Comparing and pruning defect structures across charge states...
These functions give us some info about whether any energy-lowering defect distortions were identified, and we can see the results clearer by plotting:
%matplotlib inline
figs = plotting.plot_all_defects(defect_charges_dict)
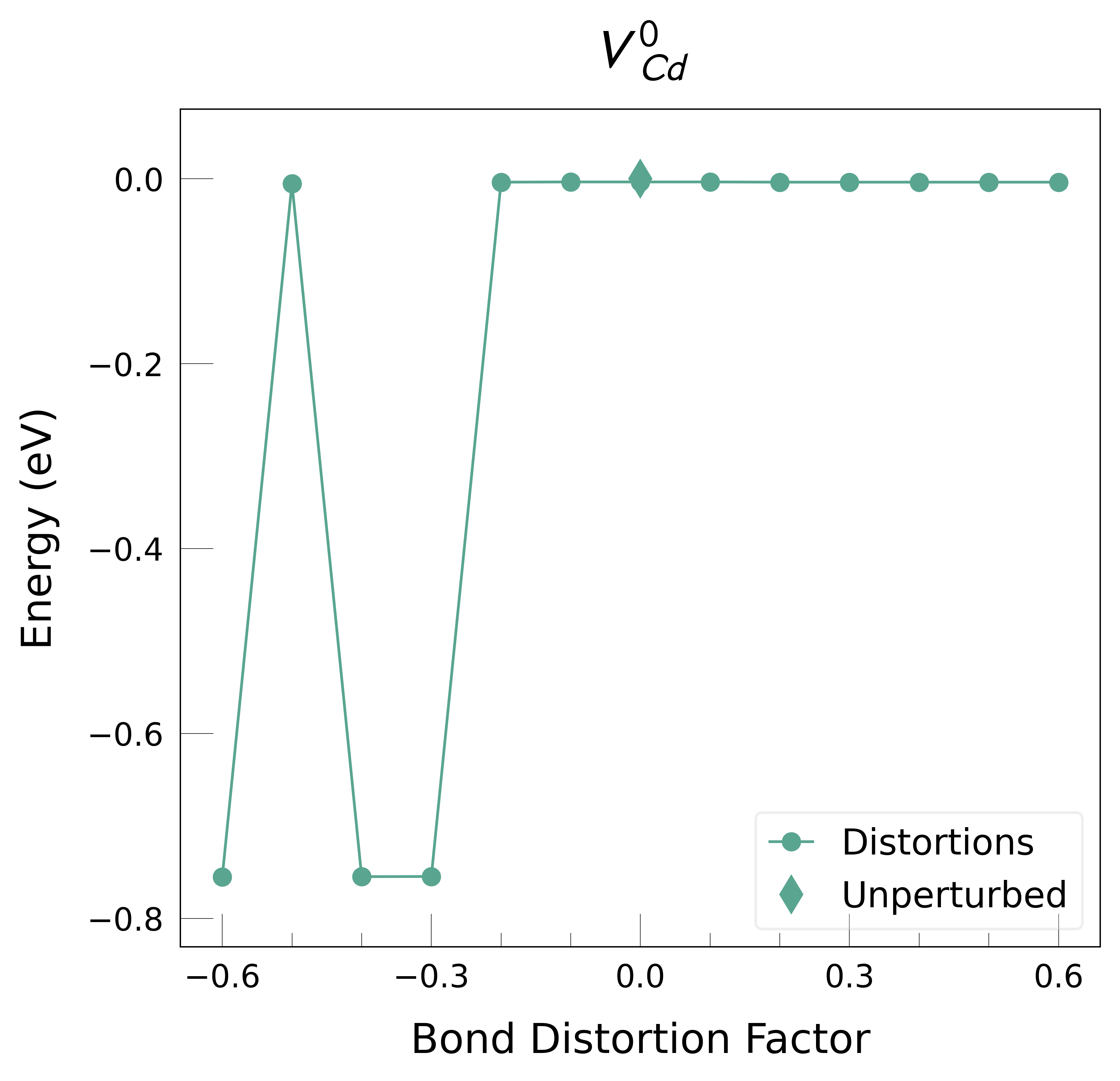
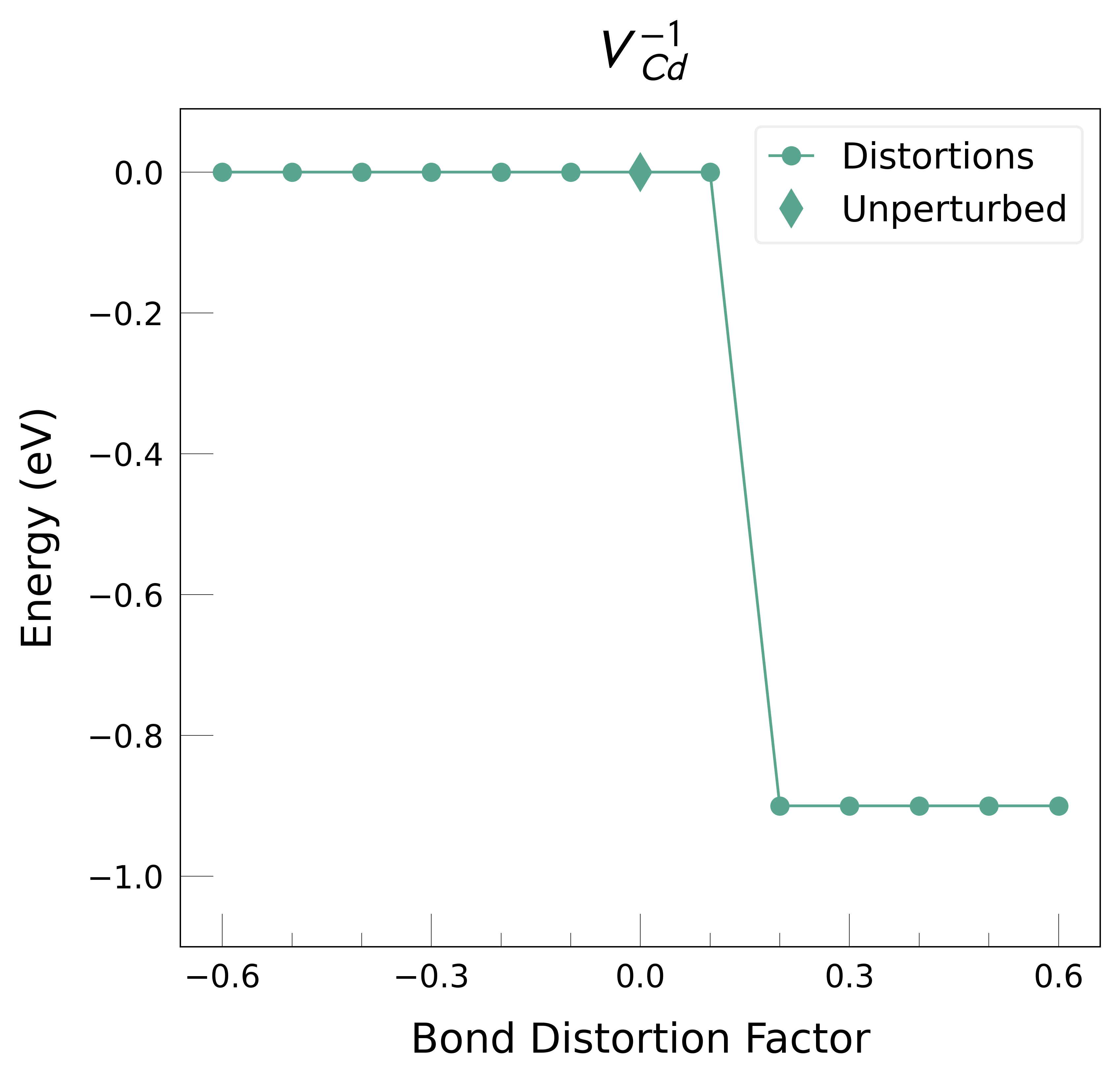
This prints the distortion plots for all defects where a significant energy lowering distortion, relative to the standard unperturbed relaxation, was identified. The threshold energy difference to consider as ‘significant’ is controlled by the min_e_diff optional parameter (default = 0.05 eV).
Can also add a colorbar#
These plots can be made more informative by adding a colorbar showing the structural similarity between the relaxed structures.
For this you need the CONTCAR’s obtained with each distortion (as mentioned above).
For the colorbar structure comparison metric, you can either use:
summed root mean squared displacement (
metric=disp)maximum distance between matching sites (
metric=max_dist, default).
figs = plotting.plot_all_defects(
defect_charges_dict,
add_colorbar=True
)
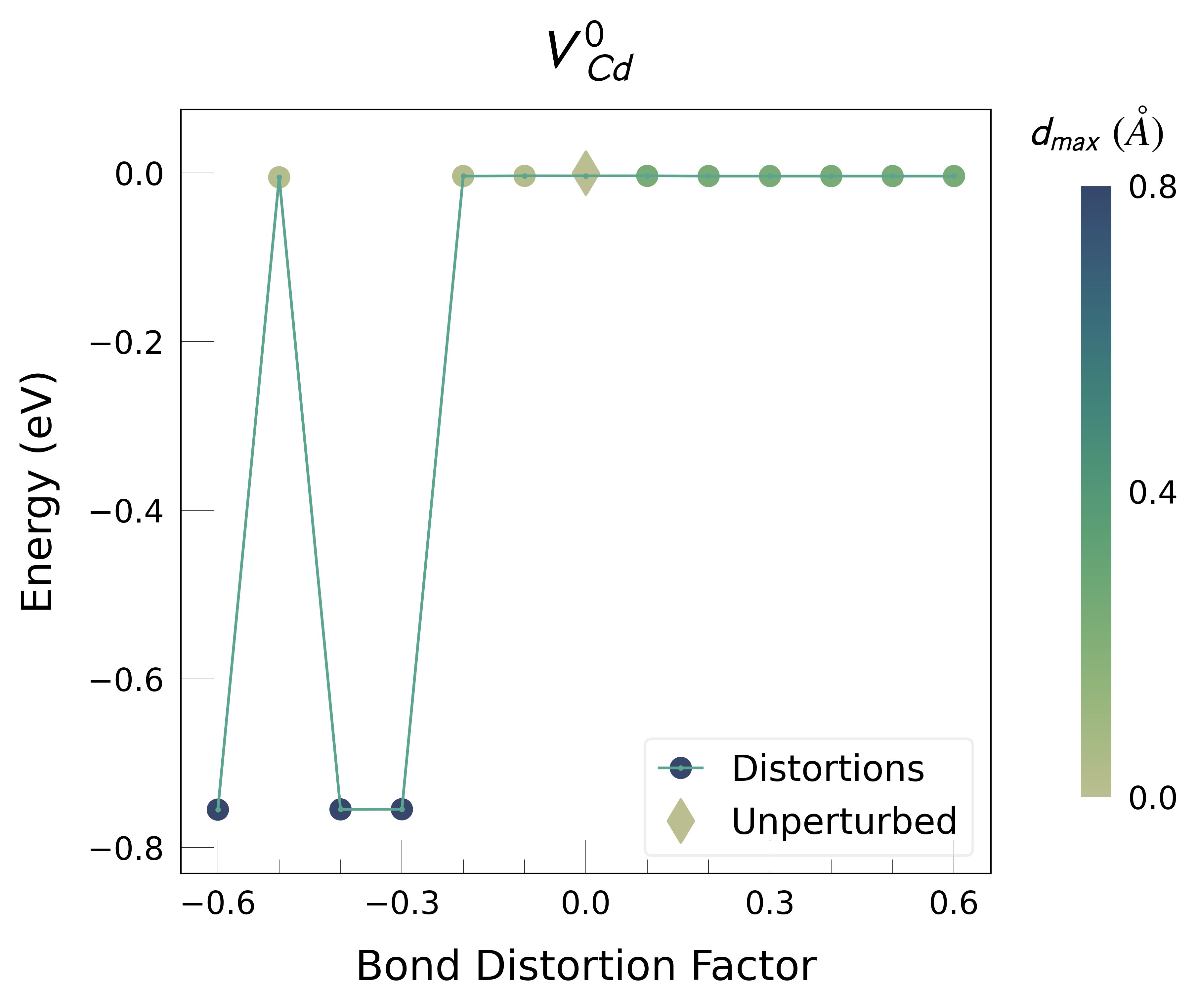
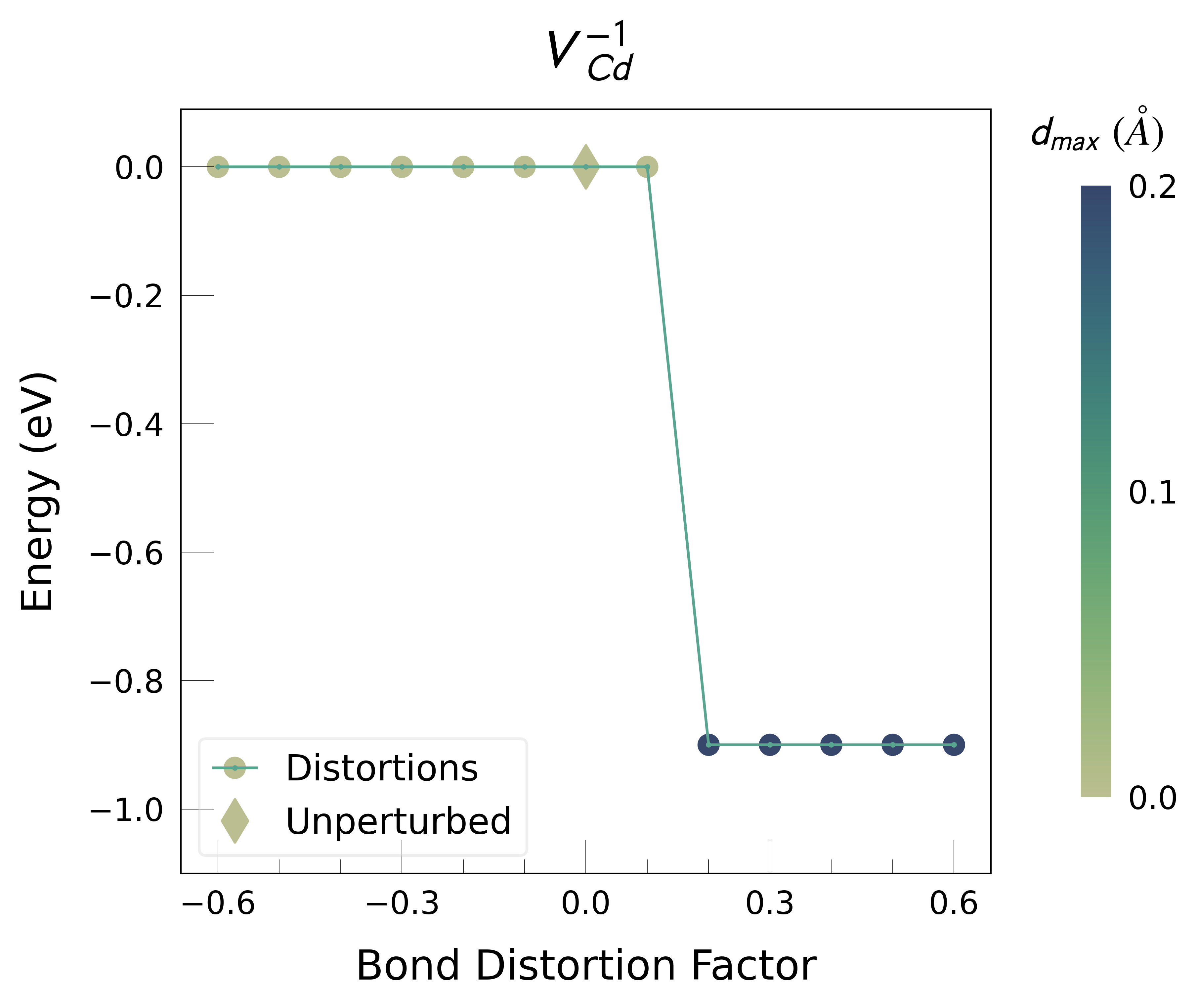
figs = plotting.plot_all_defects(
defect_charges_dict,
add_colorbar=True,
metric="disp" # customise colourbar metric
)
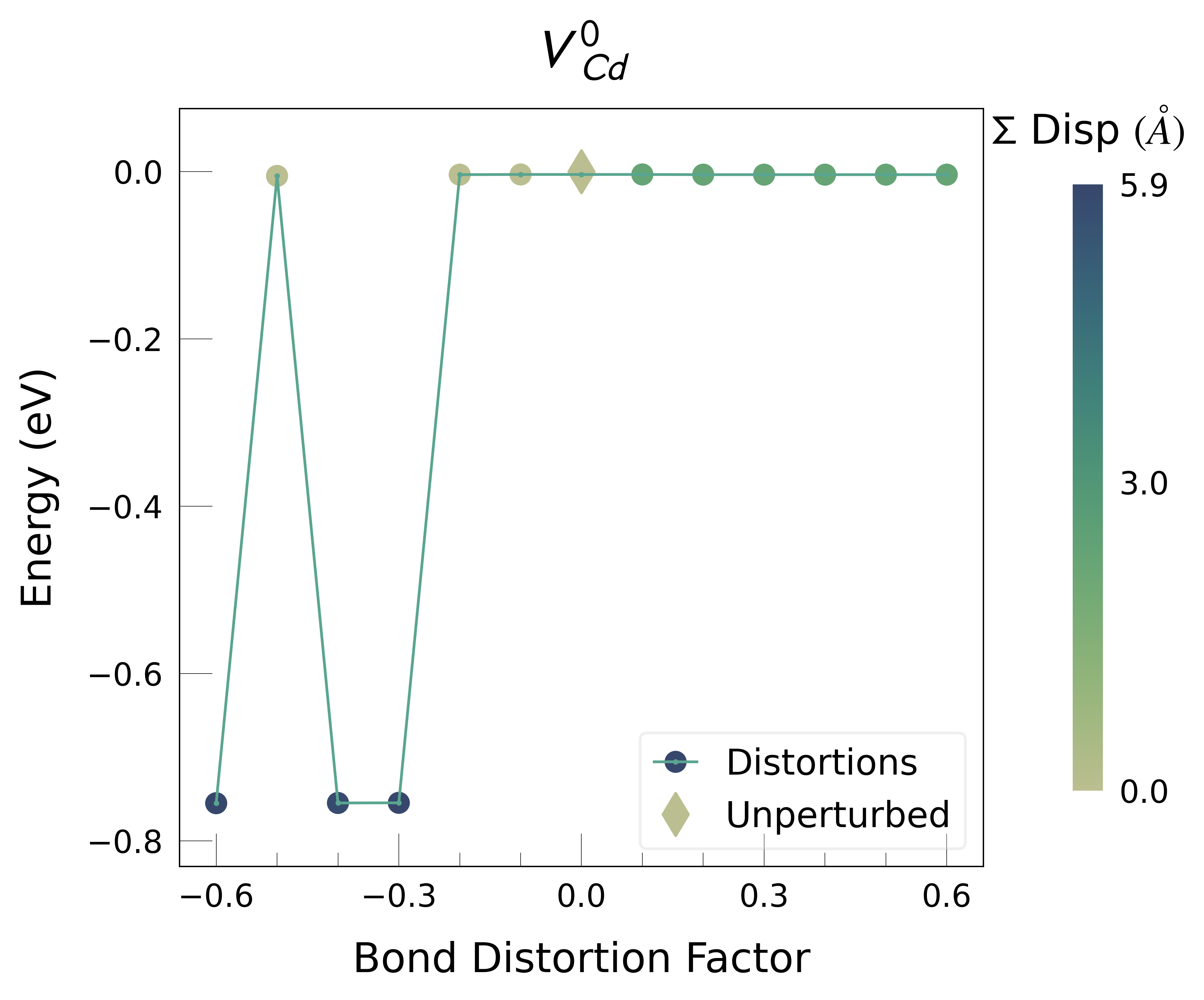
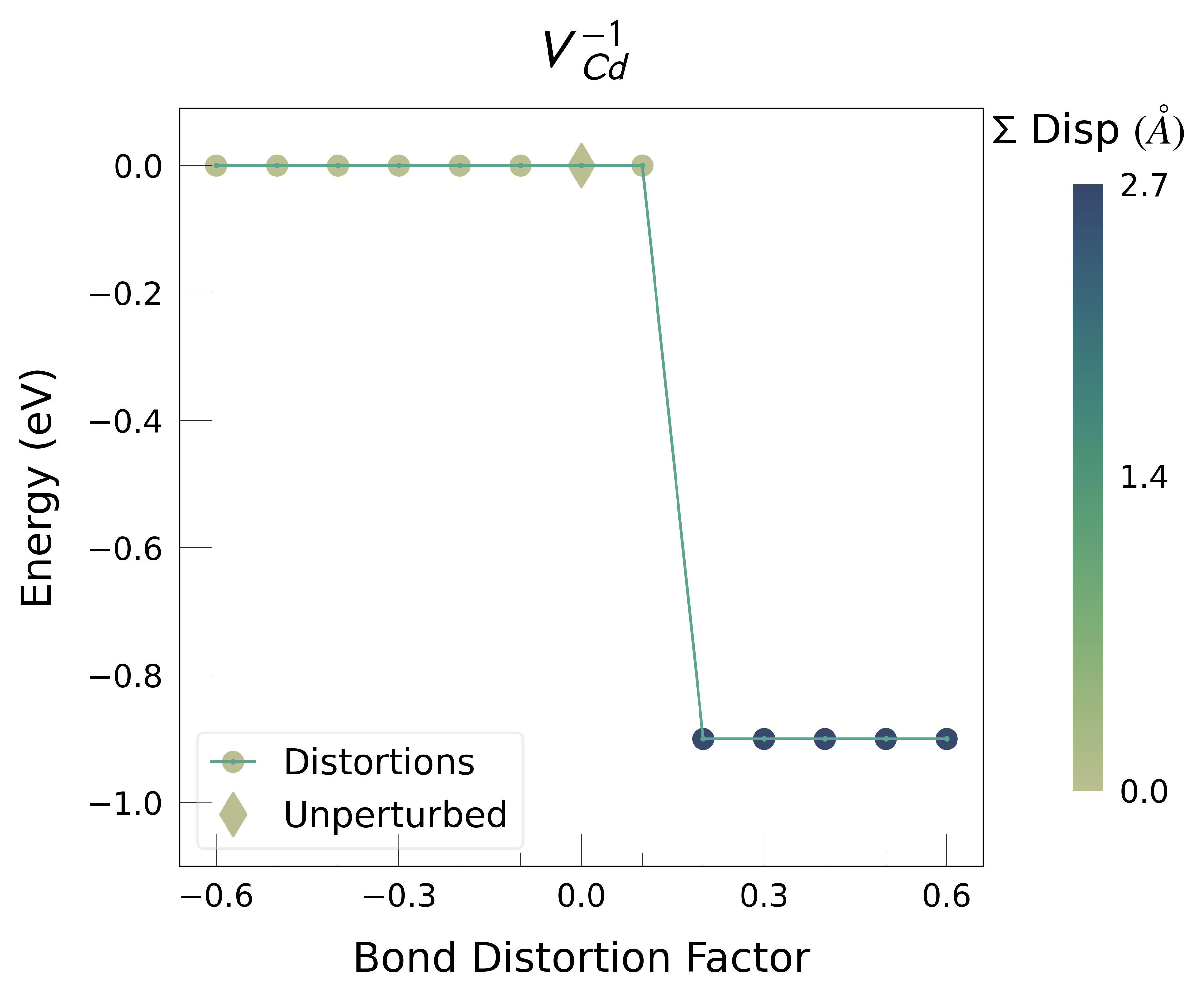
So for these example results, we find energy lowering distortions for VCd0 (at -0.3, -0.4 and -0.6 bond distortion factors) and VCd-1 (from 0.2 to 0.6 bond distortion factors). We should re-test these distorted structures for the VCd charge states where these distortions were not found, in case they also give lower energies.
Of course, this is not necessary if these structures were already found in the distortion tests for the other charge states, and so the get_energy_lowering_distortions() function automatically performs structure comparisons to determine which distortions should be tested in other charge states of the same defect, and which have already been found (see docstring for more details).
In the output of get_energy_lowering_distortions() (which we saved to low_energy_defects in the earlier cell), we get a dictionary of defects for which bond distortion found an energy-lowering distortion (which is missed with normal unperturbed relaxation), of the form {defect: [list of distortion dictionaries (with corresponding charge states, energy lowering, distortion factors, structures and charge states for which these structures weren’t found)]}.
For example, our results with VCd show that we found an energy-lowering distortion for the neutral case (subdict["charges"]) which wasn’t found with the -2 or -1 charge states (subdict["excluded_charges"]) – and so we’ll test this distorted structures with those charge states – and also an energy-lowering distortion for -1 which wasn’t found with 0 or -2 charge states.
for index, subdict in enumerate(low_energy_defects["v_Cd"]):
print(f"Energy lowering distortion number {index}")
print("Found for charge states:", subdict["charges"]) # Charge state for which the energy lowering was found
print(f"Not found in:", subdict["excluded_charges"], "\n")
Energy lowering distortion number 0
Found for charge states: [0]
Not found in: {-1, -2}
Energy lowering distortion number 1
Found for charge states: [-1]
Not found in: {0, -2}
This generates the new distorted structures and VASP inputs, to do our quick second round of structure testing (energy-lowering distortions found for at least one, but not all charge states for a given defect):
energy_lowering_distortions.write_retest_inputs(low_energy_defects)
Note
Note here the nomenclature we use for the distorted structures we’ve imported from other charge states (i.e. Bond_Distortion_-60.0%_from_0 refers to the structure obtained from relaxing the -60% distortion of the neutral (q = 0) charge state).
We can send these additional test distortions to the HPCs using this bash code:
for defect in ./*{_,_-}[0-9]/; do cd $defect; for distortion in *from*/; do scp -r ${distortion} {remote_machine}:{path to ShakeNBreak folders}/${defect}${distortion}; done; cd ..; done
If you’re using MacOS (i.e. zsh shell) you may need to run this command first for this loop to work:
setopt +o nomatch
Again we run the calculations on the HPCs, then parse and download the data to our local folders (e.g. using the same for loops above), but again in this example notebook we’ll use our fake example data for demonstration purposes as in the next cell, but don’t do this if you’re actually running the calculations!
!cp ./v_Cd_0/v_Cd_0_additional_distortions.yaml ./v_Cd_0/v_Cd_0.yaml
!cp ./v_Cd_-1/v_Cd_-1_additional_distortions.yaml ./v_Cd_-1/v_Cd_-1.yaml
!cp ./v_Cd_-2/v_Cd_-2_additional_distortions.yaml ./v_Cd_-2/v_Cd_-2.yaml
!cp ./v_Cd_0/Bond_Distortion_-60.0%/CONTCAR ./v_Cd_-1/Bond_Distortion_-60.0%_from_0/
!cp ./v_Cd_0/Bond_Distortion_-60.0%/CONTCAR ./v_Cd_-2/Bond_Distortion_-60.0%_from_0/
!cp ./v_Cd_-1/Unperturbed/CONTCAR ./v_Cd_-2/Bond_Distortion_20.0%_from_-1/
!cp ./v_Cd_-1/Unperturbed/CONTCAR ./v_Cd_0/Bond_Distortion_20.0%_from_-1/
Then re-parse with the same get_energy_lowering_distortions() function from before:
low_energy_defects = energy_lowering_distortions.get_energy_lowering_distortions(defect_charges_dict)
v_Cd
v_Cd_0: Energy difference between minimum, found with -0.6 bond distortion, and unperturbed: -0.76 eV.
Energy lowering distortion found for v_Cd with charge 0. Adding to low_energy_defects dictionary.
v_Cd_-1: Energy difference between minimum, found with -60.0%_from_0 bond distortion, and unperturbed: -1.20 eV.
Low-energy distorted structure for v_Cd_-1 already found with charge states ['0'], storing together.
v_Cd_-2: Energy difference between minimum, found with 20.0%_from_-1 bond distortion, and unperturbed: -1.90 eV.
New (according to structure matching) low-energy distorted structure found for v_Cd_-2, adding to low_energy_defects['v_Cd'] list.
Comparing and pruning defect structures across charge states...
Ground-state structure found for v_Cd with charges [0, -1] has also been found for charge state -2 (according to structure matching). Adding this charge to the corresponding entry in low_energy_defects[v_Cd].
Ground-state structure found for v_Cd with charges [-2] has also been found for charge state 0 (according to structure matching). Adding this charge to the corresponding entry in low_energy_defects[v_Cd].
Finally we can replot the results from all our distortion tests:
figs = plotting.plot_all_defects(defect_charges_dict)
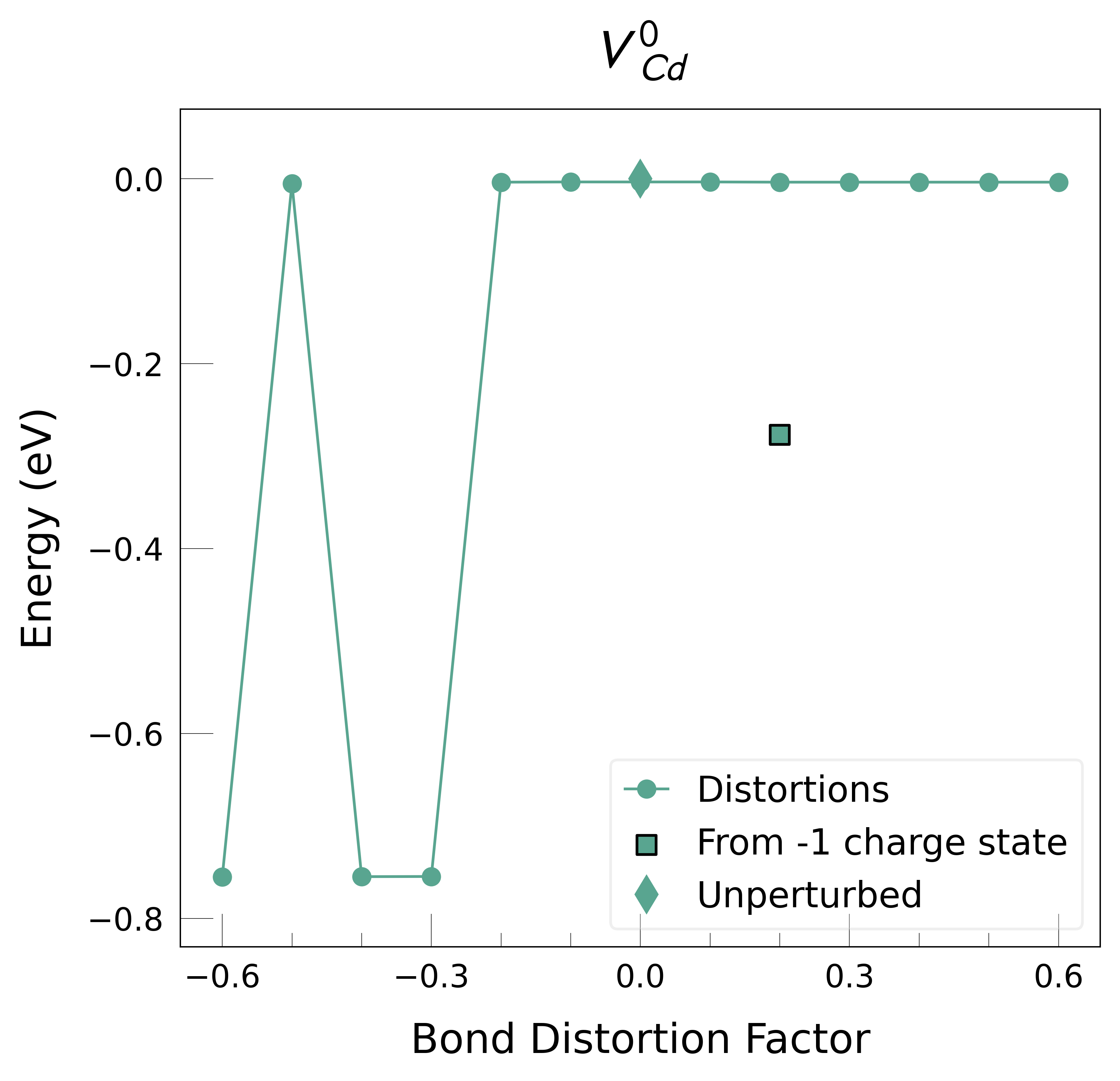
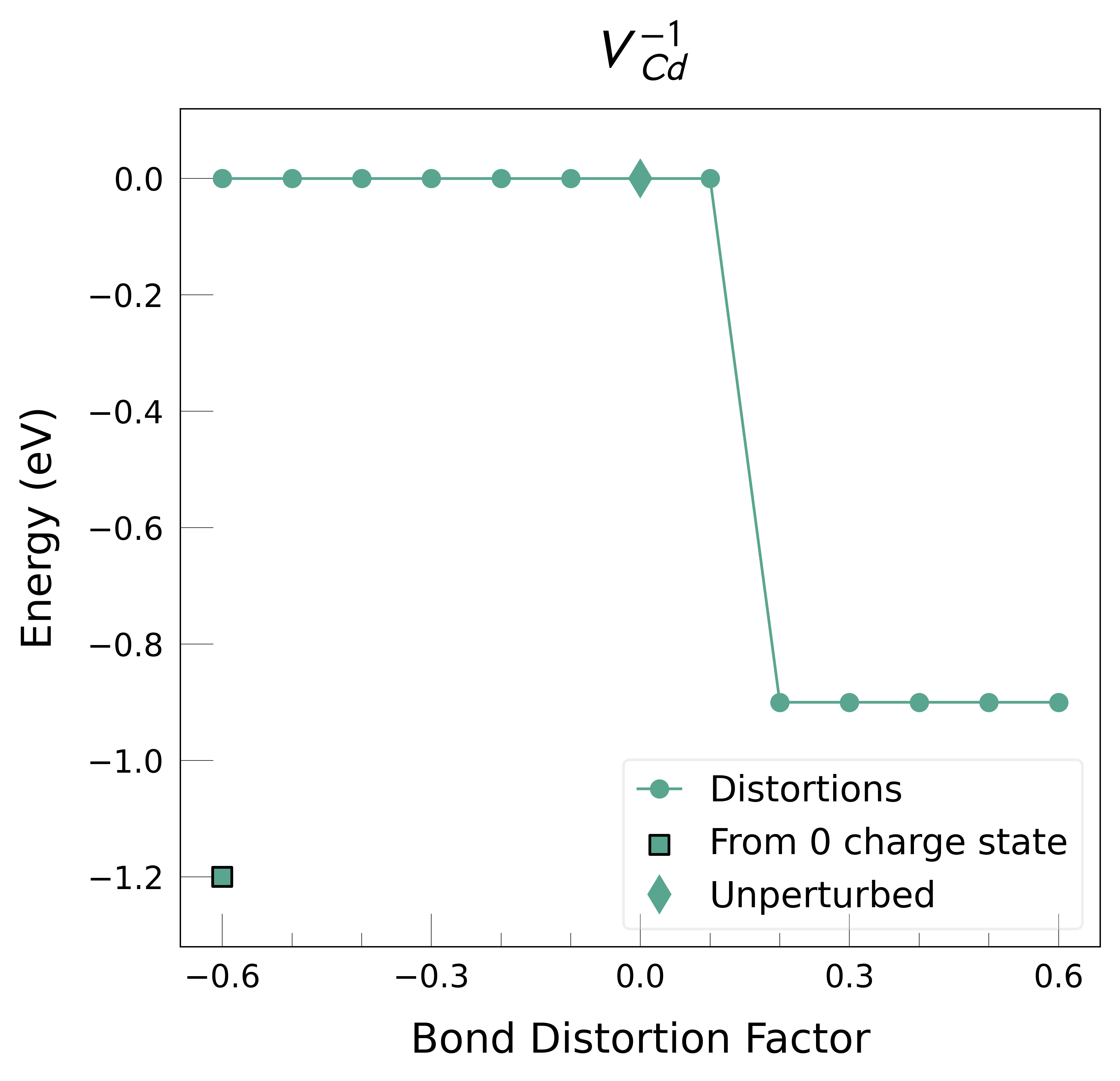
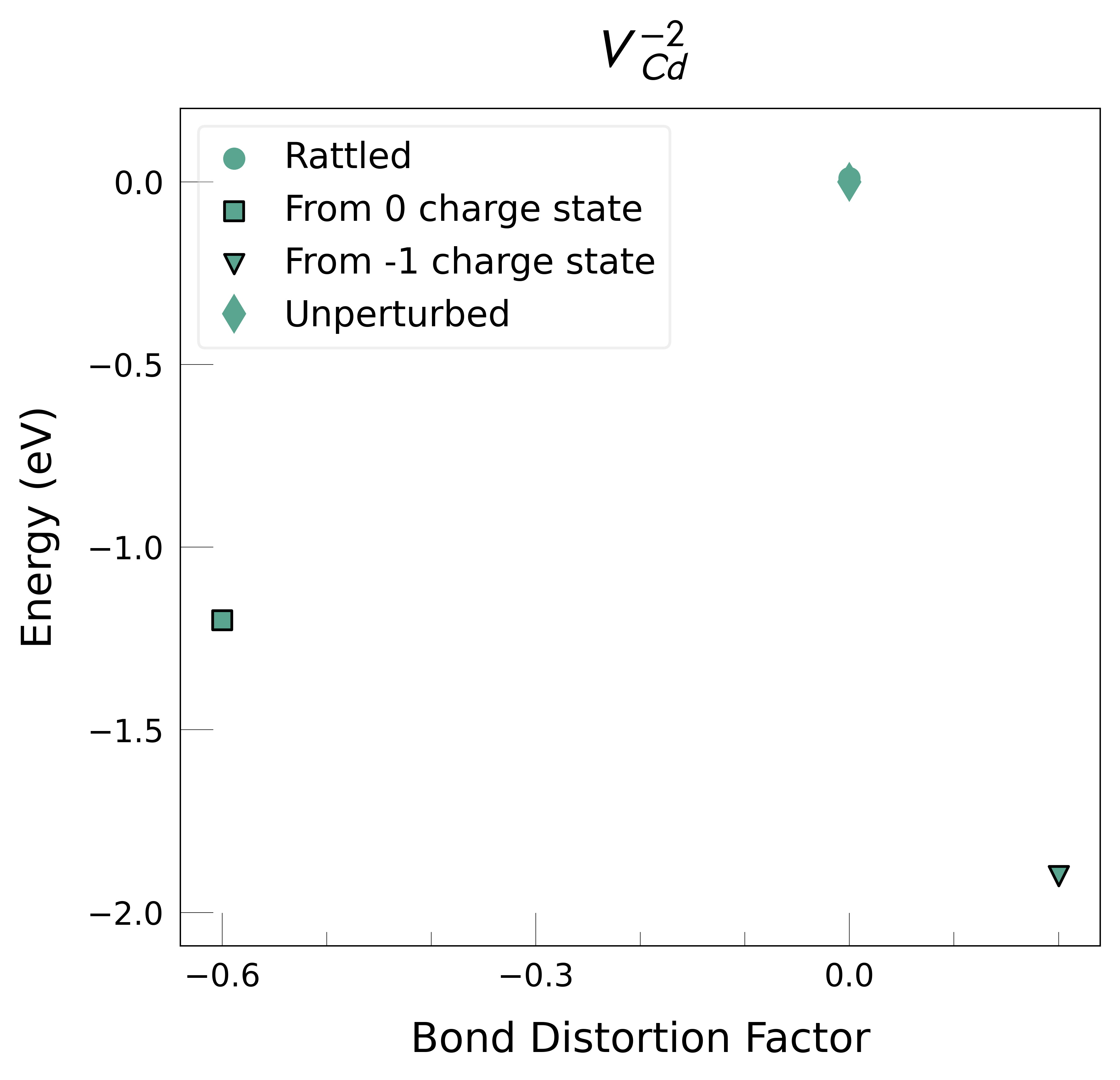
In this example case, for VCd0 the distorted structure originally found for the -1 charge state comes out lower energy than the VCd0 unperturbed relaxation, but still higher energy than the previously identified ground-state at -0.3, -0.4 and -0.6 distortion factors.
For VCd-1, the distorted structure originally found for the neutral (0) charge state comes out lower energy than the previously identified ground-state at distortion factors >0.2.
We now continue our defect calculations using the ground-state CONTCARs we’ve obtained for each defect, with our fully-converged INCAR and KPOINTS settings, to get our final defect formation energies (confident that we’ve identified the ground-state defect structure!). The energy_lowering_distortions.write_groundstate_structure() function automatically writes these lowest-energy structures to our defect folders:
energy_lowering_distortions.write_groundstate_structure(
groundstate_folder="vasp_nkred_std",
groundstate_filename="POSCAR",
)
!head v_Cd_0/vasp_nkred_std/POSCAR # groundstate structure from -60% distortion relaxation
-60.0%_Bond__vac_1_Cd[0. 0. 0.]_-dNELECT
1.0000000000000000
13.0867679999999993 0.0000000000000000 0.0000000000000000
0.0000000000000000 13.0867679999999993 0.0000000000000000
0.0000000000000000 0.0000000000000000 13.0867679999999993
Cd Te
31 32
Direct
0.0014403846070577 0.0152341826280604 0.4960600473735149
0.0018443102488570 0.5161087673464303 -0.0040398656877614
!diff v_Cd_0/vasp_nkred_std/POSCAR v_Cd_0/Bond_Distortion_-60.0%/CONTCAR # groundstate structure from -60% distortion relaxation
Tip
When using ShakeNBreak with doped for defect calculations, we would typically set groundstate_folder to vasp_nkred_std or vasp_std as shown here, to save our SnB-calculated ground-state structures to POSCAR files in these sub-directories used by default with doped, before continuing with our final fully-converged defect calculations.
5. Optional: Analyse the defect distortions found with SnB#
If we want to analyse in more detail the defect distortions identified with our structure searching, we can use some of the functions from shakenbreak.analysis:
from shakenbreak import analysis
# Parse all structures obtained with distortions and unperturbed relaxation.
# This gives a dictionary matching initial distortion to final structure
v_Cd_0 = analysis.get_structures("v_Cd_0")
# Can then analyse a chosen final structure with:
df = analysis.analyse_structure("v_Cd_0", v_Cd_0["Unperturbed"])
df = analysis.analyse_structure("v_Cd_0", v_Cd_0[-0.4])
v_Cd_0 structural analysis
Analysing site V [0. 0. 0.]
Local order parameters (i.e. resemblance to given structural motif, via CrystalNN):
| Coordination | Factor | |
|---|---|---|
| 0 | square co-planar | 0.09 |
| 1 | tetrahedral | 1.00 |
| 2 | rectangular see-saw-like | 0.01 |
| 3 | see-saw-like | 0.24 |
| 4 | trigonal pyramidal | 0.25 |
Bond-lengths (in Å) to nearest neighbours:
| Element | Distance (Å) | |
|---|---|---|
| 0 | Te | 2.60 |
| 1 | Te | 2.63 |
| 2 | Te | 2.63 |
| 3 | Te | 2.65 |
v_Cd_0 structural analysis
Analysing site V [0. 0. 0.]
Local order parameters (i.e. resemblance to given structural motif, via CrystalNN):
| Coordination | Factor | |
|---|---|---|
| 0 | square co-planar | 0.13 |
| 1 | tetrahedral | 0.74 |
| 2 | rectangular see-saw-like | 0.03 |
| 3 | see-saw-like | 0.21 |
| 4 | trigonal pyramidal | 0.21 |
Bond-lengths (in Å) to nearest neighbours:
| Element | Distance (Å) | |
|---|---|---|
| 0 | Te | 2.19 |
| 1 | Te | 2.63 |
| 2 | Te | 2.64 |
| 3 | Te | 2.30 |
We can also compare the structural similarity between all structures with compare_structures(). It prints the summed root mean squared displacement,
maximum distance between paired sites, and energy (relative to unperturbed structure) of all final structures:
defect_energies = analysis.get_energies("v_Cd_0")
structure_comparison = analysis.compare_structures(
v_Cd_0,
defect_energies
)
v_Cd_0: Energy difference between minimum, found with -0.6 bond distortion, and unperturbed: -0.76 eV.
Comparing structures to Unperturbed...
| Bond Distortion | Σ{Displacements} (Å) | Max Distance (Å) | Δ Energy (eV) | |
|---|---|---|---|---|
| 0 | -0.6 | 5.873 | 0.810 | -0.76 |
| 1 | -0.5 | 0.000 | 0.024 | -0.01 |
| 2 | -0.4 | 5.760 | 0.808 | -0.75 |
| 3 | -0.3 | 5.872 | 0.808 | -0.75 |
| 4 | -0.2 | 0.000 | 0.025 | 0.00 |
| 5 | -0.1 | 0.000 | 0.028 | 0.00 |
| 6 | 0.0 | 0.000 | 0.030 | 0.00 |
| 7 | 0.1 | 2.285 | 0.237 | 0.00 |
| 8 | 0.2 | 2.285 | 0.237 | 0.00 |
| 9 | 0.3 | 2.285 | 0.237 | 0.00 |
| 10 | 0.4 | 2.285 | 0.237 | 0.00 |
| 11 | 0.5 | 2.285 | 0.237 | 0.00 |
| 12 | 0.6 | 2.285 | 0.237 | 0.00 |
| 13 | 20.0%_from_-1 | 2.285 | 0.237 | -0.28 |
| 14 | Unperturbed | 0.000 | 0.000 | 0.00 |
Highly favourable distortions are often driven by some kind of rebonding. For most vacancies and interstitials, this entails formation of new homoionic bonds between the defect neighbours.
We can quickly check for these reconstructions using analysis.get_homoionic_bonds()
bonds = analysis.get_homoionic_bonds(
structure=v_Cd_0[-0.4], # Structure to analyse
element="Te", # we're looking for Te-Te bonds
radius=2.8, # maximum bond distance between 2 Te
verbose=False, # don't print bond distances
)
print(bonds)
print("So two of the vacancy neighbours formed a Te-Te bond to compensate for the charge deficiency")
{'Te(32)': {'Te(41)': '2.75 A'}}
So two of the vacancy neighbours formed a Te-Te bond to compensate for the charge deficiency
For defects that can result in polarons, we can check the sites with significant magnetization using analysis.get_site_magnetizations()
!cp -r ../tests/data/example_results/v_Ti_0 .
df = analysis.get_site_magnetizations(
defect_species="v_Ti_0", # neutral Ti vacancy in anatase TiO2
distortions=["Unperturbed", -0.4],
defect_site=[0.0, 0.16666666666666669, 0.25],
threshold=0.3, # to filter sites with significant magnetization
orbital_projections=False, # don't show orbital projections
)
display(df["Unperturbed"]) # 4 holes localised on 4 of the vacancy neighbours
print("So we have 4 holes localised on 4 of the oxygen ions neighbouring the vacancy")
Analysing distortion Unperturbed. Total magnetization: 4.0
Analysing distortion -0.4. Total magnetization: -0.0
No significant magnetizations found for distortion: -0.4
| Site | Frac coords | Site mag | Dist. (Å) | |
|---|---|---|---|---|
| O(35) | O(35) | [0.0, 0.167, 0.014] | 1.458 | 2.26 |
| O(53) | O(53) | [-0.0, 0.167, 0.486] | 1.478 | 2.26 |
| O(62) | O(62) | [0.165, 0.167, 0.292] | 1.522 | 1.91 |
| O(68) | O(68) | [0.835, 0.167, 0.292] | 1.521 | 1.91 |
So we have 4 holes localised on 4 of the oxygen ions neighbouring the vacancy
As printed above, no significant magnetization is found for -40.0% distortion. This configuration was found to be significantly more stable than the polaronic solution, so we can quickly use analysis.get_homoionic_bonds to see why:
bonds = analysis.get_homoionic_bonds(
structure = Structure.from_file("./v_Ti_0/Bond_Distortion_-40.0%/CONTCAR"),
element="O",
radius=2.0,
verbose=False,
)
print(bonds)
print("So the formation of an O-O bond drived this distortion")
{'O(44)': {'O(62)': '1.2 A'}}
So the formation of an O-O bond drived this distortion
See the documentation for more info and optional parameter choices etc
Further Defect Analysis#
Once the ground state (and metastable) defect structures have been identified, we will want to compute their formation energies using our final fully-converged calculation parameters (i.e. plane-wave cutoff and k-point sampling). This can be done using doped, manually (not recommended) or using the other defect codes listed on the Code Compatibility page.
As shown in the doped examples and docs, you may want to further analyse the behaviour and impact on material properties of your defects using advanced defect analysis codes such as easyunfold (to analyse the electronic structure of defects in your material), py-sc-fermi (to analyse defect concentrations, doping and Fermi level tuning), or nonrad/CarrierCapture.py (to analyse non-radiative electron-hole recombination at defects).
Note
Metastable structures can also be important to defect behaviour! This is particularly the case for defect/ion migration, electron-hole recombination at defects and non-equilibrium situations such as under illumination or ion bombardment. For example, see these papers on the impact of metastable defects in CdTe: ACS Energy Lett. 2021, 6, 4, 1392–1398 and Faraday Discuss. 2022, 239, 339-356.
In particular, symmetry-breaking as a result of structural reconstruction from the initial
(Unperturbed) high-symmetry structure can result in an increase in configurational degeneracy for
the defect, which should be accounted for when later computing concentrations and Fermi level position.
These considerations, as well as the importance of metastability and temperature effects for the free
energies (and thus concentrations) for certain defects/systems are discussed in this Tutorial Review:
Imperfections are not 0 K: free energy of point defects in crystals (Chem Soc Rev 2023) and this perspective: Guidelines for robust and reproducible point defect simulations in crystals.
6. Optional: Input File Generation for Other Codes#
a) Quantum Espresso#
We can check the arguments of the write_espresso_files method with:
Dist = Distortions(defect_gen)
Oxidation states were not explicitly set, thus have been guessed as {'Cd': 2.0, 'Te': -2.0}. If this is unreasonable you should manually set oxidation_states
Dist.write_espresso_files?
Signature:
Dist.write_espresso_files(
pseudopotentials: Optional[dict] = None,
input_parameters: Optional[str] = None,
input_file: Optional[str] = None,
write_structures_only: Optional[bool] = False,
output_path: str = '.',
verbose: Optional[bool] = False,
) -> Tuple[dict, dict]
Docstring:
Generates input files for Quantum Espresso relaxations of all output
structures.
Args:
pseudopotentials (:obj:`dict`, optional):
Dictionary matching element to pseudopotential name.
(Defaults: None)
input_parameters (:obj:`dict`, optional):
Dictionary of user Quantum Espresso input parameters, to
overwrite/update `shakenbreak` default ones (see
`SnB_input_files/qe_input.yaml`).
(Default: None)
input_file (:obj:`str`, optional):
Path to Quantum Espresso input file, to overwrite/update
`shakenbreak` default ones (see `SnB_input_files/qe_input.yaml`).
If both `input_parameters` and `input_file` are provided,
the input_parameters will be used.
write_structures_only (:obj:`bool`, optional):
Whether to only write the structure files (in CIF format)
(without calculation inputs).
(Default: False)
output_path (:obj:`str`, optional):
Path to directory in which to write distorted defect structures
and calculation inputs.
(Default is current directory: ".")
verbose (:obj:`bool`):
Whether to print distortion information (bond atoms and
distances).
(Default: False)
Returns:
:obj:`tuple`:
Tuple of dictionaries with new defects_dict (containing the
distorted structures) and defect distortion parameters.
File: ~/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/shakenbreak/input.py
Type: method
# Create an instance of Distortion class with the defect dictionary and the distortion parameters
# If distortion parameters are not specified, the default values are used
Dist = Distortions(
defects=[defect_entry],
bond_distortions=[-0.3, 0.3] # For demonstration purposes, just doing 2 distortions
)
pseudopotentials = { # Your chosen pseudopotentials
'Cd': 'Cd_pbe_v1.uspp.F.UPF',
'Te': 'Te.pbe-n-rrkjus_psl.1.0.0.UPF'}
defects_dict, distortion_metadata = Dist.write_espresso_files(
pseudopotentials=pseudopotentials,
)
Oxidation states were not explicitly set, thus have been guessed as {'Cd': 2.0, 'Te': -2.0}. If this is unreasonable you should manually set oxidation_states
Applying ShakeNBreak... Will apply the following bond distortions: ['-0.3', '0.3']. Then, will rattle with a std dev of 0.28 Å
Defect: v_Cd_s0
Number of missing electrons in neutral state: 2
Defect v_Cd_s0 in charge state: 0. Number of distorted neighbours: 2
Note
For the default coarse structure-searching input settings, either have a look at the qe_input.yaml file in the SnB_input_files folder or at the generated files.
We can modify these with the input_file or input_parameters option of the Dist.write_espresso_files method.
!cat ./v_Cd_0/Bond_Distortion_30.0%/espresso.pwi
&CONTROL
calculation = 'relax'
title = 'espresso'
nstep = 300
tstress = .false.
tprnfor = .true.
/
&SYSTEM
ibrav = 0
tot_charge = 0
ecutwfc = 30.0
nosym = .true.
occupations = 'smearing'
degauss = 0.0015
nspin = 2
input_dft = 'HSE'
exx_fraction = 0.25
starting_magnetization(1) = 0.0
starting_magnetization(2) = 0.0
ntyp = 2
nat = 63
/
&ELECTRONS
ecutwfc = 33.0
/
&IONS
/
&CELL
/
ATOMIC_SPECIES
Cd 112.414 Cd_pbe_v1.uspp.F.UPF
Te 127.6 Te.pbe-n-rrkjus_psl.1.0.0.UPF
K_POINTS automatic
1 1 1 0 0 0
CELL_PARAMETERS angstrom
13.08676800000000 0.00000000000000 0.00000000000000
0.00000000000000 13.08676800000000 0.00000000000000
0.00000000000000 0.00000000000000 13.08676800000000
ATOMIC_POSITIONS angstrom
Cd -0.1434911601 -0.2385151837 6.8150857816
Cd 0.4494288801 6.7769996959 -0.0135321750
Cd 0.1263646364 6.5320351698 6.4047505596
Cd 6.1744016994 -0.0422155671 -0.2883727639
Cd 7.0218513569 -0.1956932962 6.8368511812
Cd 6.4148946236 5.8427260793 -0.0941166578
Cd 6.6231445834 6.6386273355 6.5646951417
Cd -0.0930463691 2.7998390985 3.1815963828
Cd -0.4382789783 3.2224005685 10.1945490442
Cd 0.3283883823 9.8199609895 3.8588142250
Cd 0.2868375728 9.5008031100 10.1622309177
Cd 6.2236580741 3.3883053272 3.4377133240
Cd 6.9633774238 3.3859640414 9.6252804651
Cd 6.3916874783 9.6140830410 3.1716572177
Cd 6.4343753422 9.7039959522 9.4914454051
Cd 3.5290739650 -0.0102338238 3.5196904972
Cd 3.6622940322 0.0773629832 9.9548486402
Cd 3.6238402965 6.3664292031 3.1942466536
Cd 3.2709515176 6.5949379070 9.5272525954
Cd 10.0558411376 -0.1837771500 3.5341546226
Cd 9.6364301691 -0.0544704498 9.8810735404
Cd 9.3834495288 6.2323203692 3.2455048342
Cd 9.5864821955 6.2251964424 10.0537753778
Cd 2.9929046009 3.7695779217 0.0367936846
Cd 3.2695187347 3.4277104497 6.5121592001
Cd 3.7490142595 9.9037468136 0.4619742215
Cd 3.5005459698 9.9058044961 6.1735646663
Cd 10.0168838980 3.5064232914 -0.2220868039
Cd 10.0582554199 3.2072178847 6.4528722959
Cd 9.6639362700 9.9262589321 0.2457863270
Cd 9.9625737308 10.0083521620 6.8326782974
Te 1.4317034215 1.8330473632 5.4279434803
Te 2.1266235285 2.1266235285 10.9601444715
Te 1.9391896628 8.4788929397 4.9474309841
Te 1.6770602208 7.9814747527 11.6245303639
Te 7.8707506607 1.5517570052 4.5464792201
Te 8.4544681279 1.9570317189 11.3849977907
Te 8.3906081591 8.1873028630 4.9659486383
Te 7.6355771855 8.4896906729 11.7566752746
Te 1.9243151002 5.1586548520 1.4310602412
Te 1.6770642906 4.9734500802 8.0240863225
Te 2.1266235285 10.9601444715 2.1266235285
Te 1.2427283376 11.5044810739 8.5015890105
Te 7.9241624879 4.6167100396 1.2683696790
Te 8.3232724964 4.7490264262 8.2503126434
Te 7.7708359186 11.5137729195 1.7112542335
Te 8.4185500531 11.3625771643 8.4919150469
Te 4.9043822053 1.7707914499 1.9576447837
Te 4.7537708970 1.4950733142 7.9915316501
Te 4.8976539032 7.9146485988 1.8060744775
Te 5.0460973600 7.9200788121 8.1182771827
Te 11.7181873962 1.2522489835 1.8190296436
Te 11.7666060482 1.5075867949 8.3262988988
Te 11.8382652606 8.4193074354 1.4585392335
Te 11.2070619717 8.1626676217 8.8307785202
Te 4.7542068544 4.9329362339 5.5030757510
Te 4.7676801086 4.9457453322 11.4551487889
Te 4.9156377017 11.5432414287 4.4496059678
Te 4.4556361870 11.4784703262 11.6159865481
Te 11.6026136261 4.7068455418 5.3429055973
Te 11.6105882817 4.7319175681 11.4379375317
Te 11.6010799480 11.1856760790 5.0676572049
Te 11.8305502221 11.7838344102 10.9530493640
b) CP2K#
Dist.write_cp2k_files?
Signature:
Dist.write_cp2k_files(
input_file: Union[str, NoneType] = '/Users/skavanagh/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/shakenbreak/../SnB_input_files/cp2k_input.inp',
write_structures_only: Union[bool, NoneType] = False,
output_path: str = '.',
verbose: Union[bool, NoneType] = False,
) -> Tuple[dict, dict]
Docstring:
Generates input files for CP2K relaxations of all output structures.
Args:
input_file (:obj:`str`, optional):
Path to CP2K input file. If not set, default input file will be
used (see `shakenbreak/SnB_input_files/cp2k_input.inp`).
write_structures_only (:obj:`bool`, optional):
Whether to only write the structure files (in CIF format)
(without calculation inputs).
(Default: False)
output_path (:obj:`str`, optional):
Path to directory in which to write distorted defect structures
and calculation inputs.
(Default is current directory: ".")
verbose (:obj:`bool`, optional):
Whether to print distortion information (bond atoms and
distances).
(Default: False)
Returns:
:obj:`tuple`:
Tuple of dictionaries with new defects_dict (containing the
distorted structures) and defect distortion parameters.
File: ~/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/shakenbreak/input.py
Type: method
oxidation_states = {"Cd": +2, "Te": -2} # explicitly specify atom oxidation states
# Create an instance of Distortion class with the defect dictionary and the distortion parameters
# If distortion parameters are not specified, the default values are used
Dist = Distortions(
defects=[defect_entry],
oxidation_states=oxidation_states, # explicitly specify atom oxidation states
bond_distortions=[-0.3, 0.3] # For demonstration purposes, just doing 2 distortions
)
defects_dict, distortion_metadata = Dist.write_cp2k_files()
Applying ShakeNBreak... Will apply the following bond distortions: ['-0.3', '0.3']. Then, will rattle with a std dev of 0.28 Å
Defect: v_Cd_s0
Number of missing electrons in neutral state: 2
Defect v_Cd_s0 in charge state: 0. Number of distorted neighbours: 2
Note
And for the default coarse structure-searching input settings, either have a look at the cp2k_input.yaml file in the SnB_input_files folder or at the generated files.
We can modify these with the input_file option of the Dist.write_cp2k_files method.
!cat ./v_Cd_0/Bond_Distortion_30.0%/cp2k_input.inp
&GLOBAL
PROJECT relax ! files generated will be named relax.out etc
RUN_TYPE GEO_OPT ! geometry optimization
IOLEVEL MEDIUM ! reduce amount of IO
&END GLOBAL
&FORCE_EVAL
METHOD Quickstep
! the electronic structure part
&DFT
BASIS_SET_FILE_NAME HFX_BASIS
POTENTIAL_FILE_NAME GTH_POTENTIALS
SPIN_POLARIZED .TRUE.
CHARGE 0
&MGRID
CUTOFF [eV] 500 ! PW cutoff
&END MGRID
&QS
METHOD GPW
EPS_DEFAULT 1e-10
EXTRAPOLATION ASPC
&END QS
! use the GPW method (i.e. pseudopotential
! basedcalculations with the Gaussian and Plane
! Wavesscheme)
&DFT
&KPOINTS
SCHEME GAMMA 1 1 1 ! Gamma point only
&END KPOINTS
&END DFT
&POISSON
PERIODIC XYZ ! the default
&END POISSON
&PRINT
! at the end of the SCF procedure generate
! cubefiles of the density
&E_DENSITY_CUBE OFF
&END E_DENSITY_CUBE
&END PRINT
! use the OT METHOD for robust and efficientSCF,
! suitable for all non-metallic systems.
&SCF
SCF_GUESS RESTART ! can be used to RESTART an interrupted calculation
MAX_SCF 80
EPS_SCF 1e-06 ! accuracy of the SCF procedure typically 1.0E-6 - 1.0E-7
&OT
PRECONDITIONER FULL_SINGLE_INVERSE
MINIMIZER DIIS
&END OT
! an accurate preconditioner suitable also
! forlarger systems, the most robust choice
! (DIISmight sometimes be faster, but not
! asstable).
&OUTER_SCF ! repeat the inner SCF cycle 10 times
MAX_SCF 10
EPS_SCF 1e-06 ! must match the above
&END OUTER_SCF
! do not store the wfn
&PRINT
&RESTART
&END RESTART
&END PRINT
&END SCF
! specify the exchange and correlation treatment
&XC
! use a PBE0 functional
&XC_FUNCTIONAL
&PBE
SCALE_X 0.75
SCALE_C 1.0
&END PBE
&END XC_FUNCTIONAL
! 75% GGA exchange 100% GGA correlation
&HF
FRACTION 0.25
! 25 % HFX exchange
&SCREENING
EPS_SCHWARZ 1e-06
SCREEN_ON_INITIAL_P True
&END SCREENING
! important parameter to get stable
! HFXcalcsneeds a good (GGA) initial guess
&INTERACTION_POTENTIAL
POTENTIAL_TYPE TRUNCATED
CUTOFF_RADIUS 6.0
T_C_G_DATA ./t_c_g.dat
&END INTERACTION_POTENTIAL
! for condensed phase systemsshould be
! lessthan halve the celldata file needed with
! thetruncated operator
&MEMORY
MAX_MEMORY 4000
EPS_STORAGE_SCALING 0.1
&END MEMORY
&END HF
&END XC
&END DFT
! Description of the systemStructure will be read
! from external file
&SUBSYS
&CELL
CELL_FILE_FORMAT CIF
CELL_FILE_NAME structure.cif
&END CELL
&TOPOLOGY
COORD_FILE_NAME structure.cif
COORD_FILE_FORMAT CIF
&END TOPOLOGY
&END SUBSYS
&END FORCE_EVAL
c) CASTEP#
Dist.write_castep_files?
Signature:
Dist.write_castep_files(
input_file: Union[str, NoneType] = '/Users/skavanagh/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/shakenbreak/../SnB_input_files/castep.param',
write_structures_only: Union[bool, NoneType] = False,
output_path: str = '.',
verbose: Union[bool, NoneType] = False,
) -> Tuple[dict, dict]
Docstring:
Generates input `.cell` and `.param` files for CASTEP relaxations of
all output structures.
Args:
input_file (:obj:`str`, optional):
Path to CASTEP input (`.param`) file. If not set, default input
file will be used (see `shakenbreak/SnB_input_files/castep.param`).
write_structures_only (:obj:`bool`, optional):
Whether to only write the structure files (in CIF format)
(without calculation inputs).
(Default: False)
output_path (:obj:`str`, optional):
Path to directory in which to write distorted defect structures
and calculation inputs.
(Default is current directory: ".")
verbose (:obj:`bool`, optional):
Whether to print distortion information (bond atoms and
distances).
(Default: False)
Returns:
:obj:`tuple`:
Tuple of dictionaries with new defects_dict (containing the
distorted structures) and defect distortion parameters.
File: ~/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/shakenbreak/input.py
Type: method
oxidation_states = {"Cd": +2, "Te": -2} # explicitly specify atom oxidation states
# Create an instance of Distortion class with the defect dictionary and the distortion parameters
# If distortion parameters are not specified, the default values are used
Dist = Distortions(
defects=[defect_entry],
oxidation_states=oxidation_states, # explicitly specify atom oxidation states
bond_distortions=[0.3] # For demonstration purposes, just doing 2 distortions
)
# If we don't specify the input_file, only the structure files (in .cell format) are written
defects_dict, distortion_metadata = Dist.write_castep_files()
Applying ShakeNBreak... Will apply the following bond distortions: ['0.3']. Then, will rattle with a std dev of 0.28 Å
Defect: v_Cd_s0
Number of missing electrons in neutral state: 2
Defect v_Cd_s0 in charge state: 0. Number of distorted neighbours: 2
Note
And for the default coarse structure-searching input settings, either have a look at the castep.param file in the SnB_input_files folder or at the generated files.
We can modify these with the input_file option of the Dist.write_castep_files method.
!cat ./v_Cd_0/Bond_Distortion_30.0%/castep.param
#######################################################
#CASTEP param file: /Users/skavanagh/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/docs/v_Cd_0/Bond_Distortion_30.0%/castep.param
#Created using the Atomic Simulation Environment (ASE)#
# Internal settings of the calculator
# This can be switched off by settings
# calc._export_settings = False
# If stated, this will be automatically processed
# by ase.io.castep.read_seed()
# ASE_INTERFACE _build_missing_pspots : True
# ASE_INTERFACE _castep_command : castep
# ASE_INTERFACE _castep_pp_path : /Users/skavanagh/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/docs
# ASE_INTERFACE _check_checkfile : True
# ASE_INTERFACE _copy_pspots : False
# ASE_INTERFACE _directory : /Users/skavanagh/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/docs/v_Cd_0/Bond_Distortion_30.0%
# ASE_INTERFACE _export_settings : True
# ASE_INTERFACE _find_pspots : False
# ASE_INTERFACE _force_write : True
# ASE_INTERFACE _label : castep
# ASE_INTERFACE _link_pspots : True
# ASE_INTERFACE _pedantic : False
# ASE_INTERFACE _prepare_input_only : False
# ASE_INTERFACE _rename_existing_dir : True
# ASE_INTERFACE _set_atoms : False
# ASE_INTERFACE _track_output : False
# ASE_INTERFACE _try_reuse : False
#######################################################
GEOM_METHOD: BFGS
GEOM_CONVERGENCE_WIN: 4
GEOM_ENERGY_TOL: 0.00005 eV
GEOM_FORCE_TOL: 0.05 ev/ang
GEOM_MAX_ITER: 300
XC_FUNCTIONAL: HSE06
SMEARING_SCHEME: Gaussian
ELEC_ENERGY_TOL: 0.00005 eV
ELECTRONIC_MINIMIZER: CG
MAX_SCF_CYCLES: 50
BASIS_PRECISION: FINE
CHARGE: 0
d) FHI-aims#
Dist.write_fhi_aims_files?
Signature:
Dist.write_fhi_aims_files(
input_file: Union[str, NoneType] = None,
ase_calculator: Union[ase.calculators.aims.Aims, NoneType] = None,
write_structures_only: Union[bool, NoneType] = False,
output_path: str = '.',
verbose: Union[bool, NoneType] = False,
) -> Tuple[dict, dict]
Docstring:
Generates input geometry and control files for FHI-aims relaxations
of all output structures.
Args:
input_file (:obj:`str`, optional):
Path to FHI-aims input file, to overwrite/update
`shakenbreak` default ones.
If both `input_file` and `ase_calculator` are provided,
the ase_calculator will be used.
ase_calculator (:obj:`ase.calculators.aims.Aims`, optional):
ASE calculator object to use for FHI-aims calculations.
If not set, `shakenbreak` default values will be used.
Recommended to check these.
(Default: None)
write_structures_only (:obj:`bool`, optional):
Whether to only write the structure files (in `geometry.in`
format), (without the contro-in file).
output_path (:obj:`str`, optional):
Path to directory in which to write distorted defect structures
and calculation inputs.
(Default is current directory: ".")
verbose (:obj:`bool`, optional):
Whether to print distortion information (bond atoms and
distances).
(Default: False)
Returns:
:obj:`tuple`:
Tuple of dictionaries with new defects_dict (containing the
distorted structures) and defect distortion parameters.
File: ~/Library/CloudStorage/OneDrive-ImperialCollegeLondon/Bread/Projects/Packages/ShakeNBreak/shakenbreak/input.py
Type: method
oxidation_states = {"Cd": +2, "Te": -2} # specify atom oxidation states
# Create an instance of Distortion class with the defect dictionary and the distortion parameters
# If distortion parameters are not specified, the default values are used
Dist = Distortions(
defects=[defect_entry],
oxidation_states=oxidation_states,
bond_distortions=[0.3] # For demonstration purposes, just doing 2 distortions
)
# If we don't specify the input_file, only the structure files (in .cell format) are written
defects_dict, distortion_metadata = Dist.write_fhi_aims_files()
Applying ShakeNBreak... Will apply the following bond distortions: ['0.3']. Then, will rattle with a std dev of 0.28 Å
Defect: v_Cd_s0
Number of missing electrons in neutral state: 2
Defect v_Cd_s0 in charge state: 0. Number of distorted neighbours: 2
Note
And for the default coarse structure-searching input settings have a look at the generated files:
We can modify these with the ase_calculator or the input_file option of the Distortion.write_fhi_aims_files() method-
!cat ./v_Cd_0/Bond_Distortion_30.0%/control.in
#===============================================================================
# FHI-aims file: ./v_Cd_0/Bond_Distortion_30.0%/control.in
# Created using the Atomic Simulation Environment (ASE)
# Wed Feb 1 17:11:49 2023
#===============================================================================
k_grid 1 1 1
relax_geometry bfgs 0.005
xc hse06 0.11
hse_unit A
spin collinear
default_initial_moment 0
hybrid_xc_coeff 0.25
charge 0
#===============================================================================